

Abstract
Background: Cannabinoids (the active components of Cannabis sativa) and their derivatives have received considerable interest due to reports that they can affect the tumor growth, migration, and metastasis. Previous studies showed that the cannabinoid agonist WIN 55,212-2 (WIN) was associated with gastric cancer (GC) metastasis, but the mechanisms were unknown. Methods: The effects of WIN on GC cell migration and invasion were analyzed by the wound-healing assay and Transwell assay. Quantitative real-time PCR and Western blot were used to evaluate changes in expression of COX-2 and EMT associated markers in SGC7901 and AGS cells. Results: WIN inhibited cell migration, invasion, and epithelial to mesenchymal transition (EMT) in GC. WIN treatment resulted in the downregulation of cyclooxygenase-2 (COX-2) expression and decreased the phosphorylation of AKT, and inhibited EMT in SGC7901 cells. Decreased expression of COX-2 and vimentin, and increased expression of E-cadherin, which was induced by WIN, were normalized by overexpression of AKT, suggesting that AKT mediated, at least partially, the WIN suppressed EMT of GC cells. Conclusion: WIN can inhibit the EMT of GC cells through the downregulation of COX-2.
Introduction
Gastric cancer (GC) is one of the most common cancers worldwide [1]. The death rate caused by GC declined in the past several years due to a series of trials, resulting in therapeutic success. However, the recurrence and metastasis of GC are very common [2,3]. More than 65% of patients are treated with systemic chemotherapy as the main therapeutic option because of the unresectable nature of GC. GC cell resistance develops gradually, and is negatively correlated with the prognosis. Thus, a better understanding of the molecular mechanisms that regulate the development and progression of GC remain to be further elucidated to optimize strategies for more effective therapies.
Clinically, cannabinoids are often used to prevent some symptoms of the central nervous system, such as pain, nausea, and vomiting, in cancer patients treated with radiotherapy or chemotherapy [4]. As the principle active constituent of Cannibis sativa (marijuana), cannabinoids consist of two specific cell surface G-protein-coupled receptors. Type 1 receptor (CB1) and type 2 receptor (CB2) are important for the biological effects of cannabinoids [5]. The expression of CB1 is often found in the central nervous system, while CB2 is mainly expressed in immune systems [6].
Cannabinoids were first found to show antineoplastic effects in Lewis lung carcinoma in 1975 by Munson et al. [7]. Subsequent studies demonstrated that cannabinoids inhibit cell proliferation and induce apoptosis in many types of tumors, such as pancreatic cancer, skin cancer, lymphoma, breast cancer, and prostate cancer, by mediating various cell signaling pathways [8,9,10,11,12]. Cannabinoids also have anti-neoplastic effects in gastric cancer. WIN 55,212-2 (WIN), a cannabinoid agonist, inhibits cell proliferation by cell cycle arrest, inhibits cell invasion by downregulation of matrix metalloproteinase-2 (MMP-2) and vascular endothelial growth factor A (VEGF-A), and induces apoptosis by downregulation of phospho-AKT in gastric cancer cells [13,14]. In a murine xenograft model of gastric cancer, WIN reduced tumor growth via inducing tumor cell apoptosis [15]. A previous study showed a correlation between WIN and the metastasis of GC [13]; however, the mechanism remains unknown.
Cyclooxygenase-2 (COX-2), an isoform of COX, has pivotal roles in cellular processes such as cell proliferation, survival and apoptosis [16]. COX-2 has also been reported to be associated with EMT and metastasis of tumor cells [17,18]. Notably, previous studies have shown a positive correction between COX-2 expression and cannabidiolic acid [19]. In view of these data, we speculate that COX-2 may be one of the important downstream targets of WIN.
In the present study, we explored the biological role of WIN in GC metastasis. We found that WIN inhibited migration, invasion, and epithelial mesenchymal transition (EMT) of GC cell lines SGC7901 and AGS. WIN treatment decreased the expression levels of cyclooxygenase-2 (COX-2), p-AKT, and vimentin, and increased the expression of E-cadherin. Overexpression of AKT rescued WIN-induced altered expression of COX-2, p-AKT, vimentin, and E-cadherin in GC cells. This suggests that PI3K/AKT mediates, at least partially, the WIN suppression of EMT in GC cells. These results demonstrate that WIN inhibits the EMT of GC cells through the downregulation of COX-2.
Materials and Methods
Reagents and materials
WIN 55,212-2 was purchased from Tocris Bioscience (Bristol, UK). The primary antibodies included those for E-cadherin (E-Cad), N-cadherin (N-Cad), vimentin (Vim), fibronectin (FN), p-AKT, AKT, and GAPDH, and were purchased from Cell Signaling Technology (Danvers, MA, USA). Dimethyl sulfoxide (DMSO) and other chemicals were purchased from Sigma-Aldrich (St. Louis, MO, USA). Polymerase chain reaction (PCR) reagents were purchased from TaKaRa (Tokyo, Japan).
Cell culture and treatment
The human GC cell lines SGC7901 and AGS were purchased from the Cell Bank of Shanghai (Shanghai, China) and cultured in RPMI 1640 (Invitrogen, Carlsbad, CA, USA). All cells were supplemented with 10% fetal bovine serum (FBS), 100 units/mL penicillin, and 100 µg/mL streptomycin, and were maintained under standard cell culture conditions in a humidified 5% (v/v) atmosphere of CO2 at 37°C. WIN 55,212-2 was dissolved in DMSO for in vitro assays. Cells were treated with 5 µM of WIN 55,212-2 or equal volumes of DMSO as the vehicle control for the specified time. The final concentration of DMSO in the medium was 0.1% (v/v). At this concentration, DMSO had no effect on cell proliferation.
Transfections
The specific silencer RNAs (si-COX-2) and their negative controls (Si-NC) were purchased from Ribobio (Guangzhou, China). The human COX-2 gene was amplified from human genomic DNA. The COX-2 sequence was cloned into the pcDNA3.1 vector and verified by sequencing. The SGC7901 cells were seeded into plates and transfected with pcDNA3.1 (vector control), pcDNA3.1/COX-2, si-NC, or si-COX-2 using Lipofectamine® 2000 reagent (Invitrogen) according to the manufacturer's instructions. The final concentration in the transfection mixture was 50 nM. Three independent experiments were carried out.
In vitro migration and invasion assays
The in vitro cell migration ability was determined by the wound healing assay. AGS and SGC7901 cells, treated with WIN or untreated, were seeded into six-well plates. A standard 200 µL pipette tip was used to create an artificial wound on the confluent cell monolayer after serum starvation for 24 h. The width of the scratch gap was observed using an inverted microscope at 0 h and 24 h after the artificial wound creation. Three replicate wells were performed for each experiment. Transwell® chambers (Corning, Tewksbury, MA, USA) with an 8 µm pore size polycarbonate membrane were used to determine cell invasion according to the manufacturer's protocols. Briefly, 5 × 104 AGS or SGC7901 cells treated with WIN or untreated were suspended in serum-free media and seeded onto the upper chamber of each chamber, while the lower chamber was supplemented with media with 10% FBS as a chemoattractant. Twenty-four hours after the incubation, the cells that invaded through the membrane to the lower surface were fixed using ice-cold methanol, stained with 0.1% crystal violet solution, and counted under an inverted microscope. Five random fields were selected for analyses of each chamber. Three replicate experiments were performed for each assay.
RNA isolation and quantitative real-time PCR
Total mRNA from cultured AGS or SGC7901 cells was isolated using the TRIzol® reagent (Invitrogen) and chloroform according to the manufacturer's protocol. Five-hundred ng of total RNA was used to synthesize complementary DNA using the PrimeScript RT Reagent kit (TaKaRa). The primers were used as follows: E-cadherin, forward, 5′-TAC ACT GCC CAG GAG CCA GA-3′, reverse, 5′-TGG CAC CAG TGT CCG GAT TA-3′[20]]; N-cadherin, forward, 5′-CAG TCT TAC CGA AGG ATG TGC-3′, reverse, 5′-ATC AGC TCT CGA TCC AGA GG-3′[17]; vimentin, forward, 5′-TGA GTA CCG GAG ACA GGT GCA G-3′, reverse, 5′-TAG CAG CTT CAA CGG CAA AGT TC-3′[20]; Snail, forward, 5′-TTC TCT AGG CCC TGG CTG CTA CAA-3′, reverse, 5′-TCT CTG ACA TCT GAG TGG GTC TGG A-3′[18]; Slug, forward, 5′-CTG GTC AAG AAG CAT TTC AAC GCC-3′, reverse, 5′-AAA GAG GAG AGA GGC CAT TGG GTA-3′[19]; COX-2, forward, 5′-ATT TGA TTG ACA GTC CAC C-3′, reverse, 5′-TAC ATC ATC AGA CCA GGC A-3′[19]; and GAPDH, forward, 5′-TGA AGG TCG GAG TCA ACG GAT TTG GT-3′, reverse, 5′-CAT GTG GGC CAT GAG GTC CAC CAC-3′[20]. The mRNA expression level of GAPDH was used for normalization. All quantitative real time PCR (qRT-PCR) reactions were run using a LightCycler® 480 System II (Roche Diagnostics, Mannheim, Germany), and each experiment was performed three times. The relative expression level was measured by the 2-ΔΔCT method.
Western blot analyses
Western blot analysis was performed according to standard methods. Cells in culture were lysed in RIPA buffer (Sigma-Aldrich) along with Protease Inhibitor (Sigma-Aldrich). The lysates were then electrophoresed in a 10% SDS-PAGE gel and transferred onto polyvinylidene fluoride membranes (Millipore, Billerica, MA, USA). The membranes were blocked with 5% milk and incubated with primary antibody at 4°C overnight, followed by horseradish peroxidase (HRP)-conjugated secondary antibody incubated at room temperature for 1 h. Signals were visualized by enhanced chemiluminescence (Millipore). The protein levels were expressed relative to GAPDH levels.
Statistical analyses
The data were expressed as the mean ± SD from at least three independent experiments. The statistical significance between groups was evaluated using Student's t-test. All statistical analyses were performed using SPSS, version 18.0 statistical software (SPSS, Chicago, IL, USA). A P < 0.05 was considered statistically significant.
Results
WIN suppressed cell migration and invasion of GC cells
Previous studies have shown the crucial role of WIN in tumor cell proliferation, apoptosis, and migration [13,14,15]. In the present study, we further determined the effects of WIN on the migration and invasion of GC cells. We first measured cell migration by the wound-healing assay. As shown in Fig. 1A, 24 h after WIN treatment, the wound closures of both SGC7901 and AGS cells were promoted significantly as compared with that without WIN treatment. We also determined the influence of WIN on cell invasion of SGC7901 and AGS cells by Transwell® invasion assays, and found that the cell invasion significantly decreased after WIN treatment (Fig. 1B). These results demonstrated that WIN treatment suppressed cell migration and invasion of GC cells.

Effects of WIN 55,212-2 (WIN) on cell migration and invasion in human gastric cancer (GC) cells. (A) Cell migration was measured by the wound healing assay of SGC7901 and AGS cells treated with WIN or untreated. (B) Cell invasion was measured by the Transwell® invasion assay. *P < 0.05.
Effects of WIN 55,212-2 (WIN) on cell migration and invasion in human gastric cancer (GC) cells. (A) Cell migration was measured by the wound healing assay of SGC7901 and AGS cells treated with WIN or untreated. (B) Cell invasion was measured by the Transwell® invasion assay. *P < 0.05.
WIN inhibited the EMT of GC cells
EMT allows cancer cells to acquire the mesenchymal cell phenotype from the epithelial cell phenotype, and further enhances migratory capacity and invasiveness [17]. We determined the role of WIN in the EMT of GC cells using qRT-PCR and western blots. The results shown in Fig. 2A indicate that the epithelial marker E-cadherin increased, while the mesenchymal markers Snail, Slug, vimentin, and N-cadherin decreased significantly at the mRNA levels in SGC7901 and AGS cells treated with WIN. At the protein levels, E-cadherin showed an increased expression, while Snail, Slug, vimentin, and N-cadherin showed decreased expression in both SGC7901 and AGS cells after WIN treatment (Fig. 2B). These observations suggested that WIN plays an important role in inhibiting EMT of GC cells.
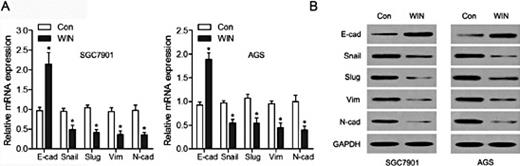
WIN inhibited endothelial to mesenchymal transition (EMT) of GC cells. (A) The mRNA levels of E-cadherin, Snail, Slug, vimentin, and N-cadherin were analyzed by quantitative real time polymerase chain reaction (qRT-PCR) in SGC7901 and AGS cells treated with WIN. (B) The protein levels of E-cadherin, Snail, Slug, vimentin, and N-cadherin, were measured by western blot analysis in SGC7901 and AGS cells treated with or without WIN. *P < 0.05.
WIN inhibited endothelial to mesenchymal transition (EMT) of GC cells. (A) The mRNA levels of E-cadherin, Snail, Slug, vimentin, and N-cadherin were analyzed by quantitative real time polymerase chain reaction (qRT-PCR) in SGC7901 and AGS cells treated with WIN. (B) The protein levels of E-cadherin, Snail, Slug, vimentin, and N-cadherin, were measured by western blot analysis in SGC7901 and AGS cells treated with or without WIN. *P < 0.05.
WIN significantly downregulated COX-2 in GC cells
We next measured the expression levels of COX-2 in GC cells and the role of WIN in COX-2 expression. The expression levels of COX-2 were detected in SGC7901 and AGS cells which were treated with WIN for 0 h, 10 h, 20 h, and 40 h. As shown in Fig. 3A, COX-2 mRNA expression levels in both SGC7901 and AGS cells decreased gradually with time of WIN treatment. The protein expression levels of COX-2 showed the same tendency (Fig. 3B), indicating that WIN inhibits COX-2 expression of GC cells in a time-dependent manner.

WIN decreased the cyclooxygenase-2 (COX-2) expression in GC cells. SGC7901 and AGS cells were treated with or without WIN for the indicated times (h = hours), then the mRNA and protein levels of COX-2 were measured using qRT-PCR (A) and western blots (B), respectively. *P < 0.05.
WIN decreased the cyclooxygenase-2 (COX-2) expression in GC cells. SGC7901 and AGS cells were treated with or without WIN for the indicated times (h = hours), then the mRNA and protein levels of COX-2 were measured using qRT-PCR (A) and western blots (B), respectively. *P < 0.05.
COX-2 was involved in WIN-induced EMT suppression
To explore the role of COX-2 in the process of WIN inhibition of EMT, COX-2 was silenced by the specific silencer RNA (si-COX-2) and then transfected into SGC7901 cells, while si-NC transfected SGC7901 cells served as the control. As shown in Fig. 4A, expression of the epithelial marker E-cadherin in SGC7901 cells increased, while the expression levels of the mesenchymal markers vimentin and N-cadherin decreased significantly after COX-2 silencing, suggesting that silencing of COX-2 inhibits EMT. We then transfected SGC7901 cells with pcDNA3.1 (vector) as the control or with pcDNA3.1/COX-2 for the constitutive activation of COX-2 (Fig. 4B). COX-2 overexpression significantly promoted EMT by downregulation of vimentin and upregulation of E-cadherin, whereas WIN attenuated these effects (Fig. 4C). The role of COX-2 in the WIN inhibition of EMT was also confirmed by morphological studies that WIN suppressed COX-2-induced mesenchymal morphological changes.

COX-2 was involved in WIN-induced EMT inhibition in SGC7901 cells. (A) SGC7901 cells were transfected with si-NC or si-COX-2, and then the protein expression of COX-2, E-cadherin, vimentin, and N-cadherin was analyzed by western blots. (B) COX-2 expression was determined by western blots of SGC7901 cells transfected with pcDNA3.1 (vector) or pcDNA3.1/COX-2 plasmid. (C) Transfected cells were further treated with WIN and then the expression of E-cadherin and vimentin were determined by western blots. (D) Cell morphological changes were observed with phase-contrast microscopy.
COX-2 was involved in WIN-induced EMT inhibition in SGC7901 cells. (A) SGC7901 cells were transfected with si-NC or si-COX-2, and then the protein expression of COX-2, E-cadherin, vimentin, and N-cadherin was analyzed by western blots. (B) COX-2 expression was determined by western blots of SGC7901 cells transfected with pcDNA3.1 (vector) or pcDNA3.1/COX-2 plasmid. (C) Transfected cells were further treated with WIN and then the expression of E-cadherin and vimentin were determined by western blots. (D) Cell morphological changes were observed with phase-contrast microscopy.
The activation of PI3K/AKT was involved in WIN-induced COX-2 downregulation
It has been reported that WIN induces the activation of the PI3K/AKT signaling pathway in cancer cells [21]. We further wished to determine whether the PI3K/AKT signaling pathway played an important role in the process of WIN-induced COX-2 downregulation. As shown in Fig. 5A, WIN treatment resulted in the downregulation of COX-2 and p-AKT, and in the upregulation of E-cad in SGC7901 cells. The cells were transfected with pcDNA3.1/AKT plasmid for the constitutive activation of AKT, while transfection with pcDNA3.1 (vector) was used as a control. The results shown in Fig. 5A demonstrated that WIN significantly decreased the phosphorylation of AKT in SGC7901 cells. Overexpression of AKT significantly increased COX-2 expression and attenuated WIN-induced EMT suppression (Fig. 5B). Together, these data suggested that the PI3K/AKT signaling pathway mediates, at least partially, the WIN-induced COX-2 downregulation and the EMT of GC cells.

PI3K/AKT mediated the induction effects of WIN on COX-2 downregulation and EMT upregulation in GC cells. (A) Cells were treated with WIN, and the phosphorylated AKT level was measured by western blots. (B) Cells were transfected with pcDNA3.1 (vector) or pcDNA3.1/AKT plasmid, further treated with WIN, and the expression of COX-2, E-Cadherin, and vimentin levels were determined by western blots. *P < 0.05.
PI3K/AKT mediated the induction effects of WIN on COX-2 downregulation and EMT upregulation in GC cells. (A) Cells were treated with WIN, and the phosphorylated AKT level was measured by western blots. (B) Cells were transfected with pcDNA3.1 (vector) or pcDNA3.1/AKT plasmid, further treated with WIN, and the expression of COX-2, E-Cadherin, and vimentin levels were determined by western blots. *P < 0.05.
Discussion
GC generally results in an aggressive clinical course, especially because of the metastasis of tumor cells to distant organs [22]. EMT has been proven to play a crucial role in the process of metastasis [23]. It is therefore beneficial for patients with gastric cancer to develop new drugs which inhibit the process of EMT.
Previous studies have shown that the cannabinoid agonist WIN 55,212-2 (WIN) reduces cell proliferation, migration, and invasion, induces apoptosis, and G0/G1 cell cycle arrest of GC cells [24,25]. However, the role of WIN in GC cell metastasis and its molecular mechanism remain to be explored. In the present study, we explored the biological role of WIN in GC metastasis. WIN treatment inhibited migration and invasion of GC cells, which is consistent with previous studies [26]. More importantly, WIN treatment showed an obvious inhibition of EMT in GC cells, which was verified by morphological changes and by qRT-PCR and western blots. EMT promotes metastasis, which causes about 90% of death of patients with cancer, indicating that WIN has the potential for clinical use as a drug against metastasis.
When exposed at the invasive front of a carcinoma, epithelial cells often acquire an enhanced migratory phenotype, which is defined as EMT. During the process of metastasis of various cancers, EMT promotes the changes of morphology and function of epithelial cells to mesenchymal cells [27]. There are some markers used to detect the status of these cells. E-cadherin is often considered to be the epithelial marker, while Snail, Slug, vimentin, and N-cadherin are considered mesenchymal markers. In our present study, we treated AGS cells and SGC7901 cells with WIN, and then detected the expression levels of EMT markers at the mRNA and protein levels. We found that WIN treatment promoted epithelial marker and inhibited mesenchymal marker expressions in both of the GC cells.
COX-2 is induced by a variety of factors. It catalyzes the metabolism of unsaturated fatty acids into short-lived 15-hydroperoxide prostaglandin G2 (PGG2), and is involved in the first two steps of the synthesis of prostanoids [28]. COX-2 is also reported to be associated with carcinogenesis, promoting proliferation, invasion, angiogenesis, and metastasis [29]. In various tumors, such as lung cancer, pancreatic cancer, colorectal cancer, breast cancer, and liver cancer, COX-2 was found to be overexpressed [30,31,32,33,34,35]. In the present study, we showed that WIN treatment inhibited GC invasion. We further explored the role of WIN on COX-2 expression, which has been shown to be involved in tumor progression. Our results suggested that WIN treatment inhibited COX-2 expression in a time-dependent manner. Inhibition of COX-2 expression in SGC7901 cells by a specific silencer RNA resulted in the upregulation of an epithelial marker, in the downregulation of mesenchymal markers, and in morphological changes, while overexpression of COX-2 by pcDNA3.1/COX-2 resulted in the opposite effects on EMT, indicating the important role of COX-2 in EMT.
The PI3K/AKT signaling pathway plays a crucial role in various biological processes, such as cell proliferation, migration, invasion, and metastasis, and is often deregulated in various cancers, to accordingly serve as an important anticancer target [36]. Our results showed that WIN treatment inhibited p-AKT expression in SGC7901 cells, along with the downregulation of COX-2. Moreover, overexpression of AKT resulted in the upregulation of COX-2, indicating that the PI3K/AKT signaling pathway was involved in the process of WIN induced COX-2 downregulation.
Our objective was to explore the potential for the clinical application of WIN in GC therapy. We have shown the potential of WIN in GC cell lines; however, there are no WIN studies in GC mouse models. Future research should focus on the role of WIN in GC mouse models to verify the results using human GC clinical samples.
In summary, we found that WIN inhibited cell migration, invasion, and EMT of GC cells through downregulation of COX-2. These results provide new insights into the underlying mechanisms of WIN on GC cell invasion and EMT, and may contribute to the design of novel therapeutic strategies for tumor inhibition. Further studies are needed to demonstrate the effects of WIN on GC metastasis in vivo.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (81402488).
Disclosure Statement
The authors declare to have no conflicts of interest.

