

Abstract
Background/Aims: Abnormal proliferation of vascular smooth muscle cells (VSMCs) is a hallmark of vascular lesions, such as atherosclerosis and restenosis. PDGF-ββ, an isoform of PDGF (platelet-derived growth factor), has been demonstrated to induce proliferation and migration of VSMCs. Atorvastatin calcium, a selective inhibitor of 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase, has favorable protective effects on VSMCs. This study examined the effects of atorvastatin calcium on the proliferation and migration of PDGF-ββ-treated VSMCs, as well as its underlying mechanisms. Methods: MTT assays, Edu imaging, cell cycle analysis, wound healing assays, transwell migration assays, and western blot analysis were performed. Results: Atorvastatin calcium significantly inhibited cell proliferation, DNA synthesis and cell migration of PDGF-ββ-treated VSMCs. We demonstrated that atorvastatin calcium induced cell cycle arrest in the G0/G1 phase in response to PDGF-ββ stimulation and decreased the expression of G0/G1-specific regulatory proteins, including proliferating cell nuclear antigen (PCNA), CDK2, cyclin D1, cyclin E and CDK4 in PDGF-ββ-treated VSMCs. Moreover, pretreatment with atorvastatin calcium inhibited the PDGF-ββ-treated phosphorylation of PDGFRβ and Akt, whereas atorvastatin calcium did not affect the phosphorylation of PLC-γ1 or (ERK) 1/2. Conclusion: Our data suggested that atorvastatin calcium inhibited abnormal proliferation and migration of VSMCs through G0/G1 cell cycle arrest and suppression of the PDGFRβ-Akt signaling cascade.
Introduction
Abnormal proliferation and migration of vascular smooth muscle cells (VSMCs) play important roles in the development of vascular disorders, such as atherosclerosis, neointima formation and restenosis [1, 2]. VSMCs are an important component of the media layer of mature blood vessels and have contractile characteristics to regulate blood vessel tone [3]. VSMCs normally exist in a non-proliferative quiescent state and express a series of contractile proteins, exhibiting differentiated and contractile phenotypes [4]. However, after vascular injury, including atherosclerosis, restenosis after angioplasty, stenting, or bypass surgery, VSMCs dedifferentiate to a synthetic phenotype and re-enter the cell cycle and undergo abnormal proliferation and migration [1, 5-7]. The synthetic phenotype VSMCs lead to increased proliferation, migration and synthesis of extracellular matrix proteins, leading to intimal hyperplasia [2, 8, 9].
Platelet-derived growth factor (PDGF), as one of the most potent mitogens and chemoattractants for VSMCs, is produced by activated macrophages, VSMCs, and endothelial cells, and PDGF plays an essential role in abnormal proliferation and migration of VSMCs [10]. The PDGF family includes PDGF-AA, PDGF-ββ, PDGF-Aβ, PDGF-CC, and PDGF-DD. Among the isoforms of the PDGF family, PDGF-ββ-treated proliferation of VSMCs has been well demonstrated [11, 12]. The binding of PDGF-ββ to the PDGF receptor (PDGF-R) leads to the phosphorylation of PDGF-Rβ-chain (PDGF-Rβ] tyrosine residues and then activates many downstream signaling molecules, such as phosphatidylinositol 3-kinase (PI3K)/Akt, phospholipase C (PLC)-γ1 and extracellular signal regulated kinase (ERK) 1/2 pathways [13-15]. It has been shown that Akt is a downstream target of PI3K and that both Akt and ERK1/2 signaling molecules play essential roles in cell proliferation, migration and other mammalian cellular processes [16-18]. Activation of these signaling pathways can lead to the progression and development of proliferative vascular diseases, including atherosclerosis and hypertension [18-20]. Therefore, modulation of the related signaling pathways may be a vital pharmacological point for the prevention of cardiovascular diseases.
Atorvastatin calcium, a 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitor, has been widely used in clinical patients because of its lipid-reducing effect. Moreover, statins have also been reported to have a broad spectrum of biological activities, such as anti-oxidation, anti-inflammation and anti-thrombosis [21-24]. Although studies have demonstrated that statins can reduce intimal hyperplasia and slow atherosclerosis progression via inhibiting VSMCs proliferation and migration [25], the mechanisms behind the favorable effects of atorvastatin calcium on VSMCs are still not fully understood.
In this study, we examined the anti-proliferation and anti-migration effects of atorvastatin calcium on PDGF-ββ-treated VSMCs. Furthermore, we evaluated the effects of atorvastatin calcium on PDGF-ββ-treated cell cycle progression in VSMCs and on cell cycle regulatory proteins including cyclins and CDKs, as part of the early G0/G1 interphase transition. In addition, we also determined the effect of torvastatin calcium on the expression of migration-related proteins in PDGF-ββ-treated VSMCs. Finally, we investigated atorvastatin calcium’s influence on PDGF-Rβ, PI3K/Akt, PLC-γ1, and ERK1/2 pathways in PDGF-ββ-treated VSMCs.
Materials and Methods
Ethics Statement
All experimental protocols were approved by the Ethics Committee of China Medical University (Shenyang, China). All procedures were carried out in accordance with the ethical standards and the Guide for the Care and Use of Laboratory. Male Sprague-Dawley rats, 8 weeks, weighing 150–200 g were placed under general anesthesia with pentobarbital sodium (50 mg/kg) to harvest tissues for VSMC isolation as described previously [26], and all efforts were made to minimize suffering.
Isolation and culture of VSMCs
The VSMCs were isolated from male Sprague-Dawley rats as described previously [26] and cultured in DMEM (Thermo Fisher, Shanghai, China) supplemented with 10% fetal bovine serum (FBS; Thermo Fisher, Shanghai, China) and 1% antibiotics at 37.0°C in a humidified 5% CO2 incubator. VSMCs from passages 4-7 were used in this experiment. The VSMCs were cultured for 48 h and serum-starved for 24 h before each experiment.
Cell proliferation assay and DNA Synthesis
Cell proliferation and DNA synthesis were determined as previously described [26]. In brief, for direct cell counting, VSMCs were seeded into 12-well culture plates at 4×104 cells/ml and then cultured in DMEM containing 10% FBS at 37.0°C until 70~80% cell confluence, and the cells were incubated with serum-free medium for 24 h and treated with various concentrations of atorvastatin calcium (dissolved in methanol) for 30 min in fresh medium and then stimulated by PDGF-ββ (20 ng/ml) for 24 h. The cells were trypsinized and counted using a hemocytometer under microscopy.
For the MTT assay, cells were seeded in 96-well culture plates (5000/well) and incubated with 0.5 mg/mL MTT in the last 4 h of the culture period at 37°C. The reaction was terminated by incubating the cells with DMSO for 10 min. An automatic microplate reader microplate reader (Bio-Rad, Hercules, CA, USA) was used to determine the absorbance at 570 nm.
DNA synthesis was performed by a 5-ethynyl-2´-deoxyuridine (Edu) incorporation assay (Click-iT® EdU Imaging Kits, Invitrogen, USA) according to the manufacturer’s instructions. Briefly, cells were incubated with EdU-labeling solution for 2 h at 37°C, and then the cells were fixed cells with 4% cold formaldehyde for 30 min at room temperature. After permeabilization with 1% Triton X-100, the cells were reacted with Click-iT® reaction cocktails (Invitrogen) for 30 min. Subsequently, the DNA contents of the cells were stained with Hoechst 33342 for 30 min. Finally, EdU-labeled cells were counted using fluorescence microscopy (CKX41-F32FL, Olympus) and normalized to the total number of Hoechst-stained cells.
Cell cycle progression analysis
VSMCs were seeded into 6-well culture plates at 1 × 105 cells/mL and then cultured in DMEM containing 10% FBS at 37°C for 24 h until 70~80% confluence. The medium was then replaced with serum-free media for 24 h. Cells were stimulated with 20 ng/mL PDGF-ββ with or without atorvastatin calcium for 24 h, trypsinized, and then centrifuged at 1500 × g for 10 min. The resulting pellets were suspended in 1 mL of 1 × PBS, washed twice, and re-centrifuged. The pellets were suspended in 70% ethanol and fixed overnight at 4°C. The fixed VSMCs were briefly vortexed and centrifuged at 15, 000 × g for 5 min. The ethanol was discarded and the pellets were stained with 0.4 mL of propidium iodide (PI) solution (50 µg/mL PI in buffer containing 100 µg/mL of RNase A). Samples were incubated for 1 h at room temperature before analysis by flow cytometry. The PI-DNA complex in each cell nucleus was measured using a FACScan instrument (Becton Dickinson, IN, United States). The proportions of cells in G0/G1, S and G2/M phases were determined using the computer program ModFitLT (Verity Software House, Topsham, ME, USA).
Wound healing assay
VSMCs were seeded into 6-well culture plates until 70~80% confluence. The VSMCs were cultured with serum-free media for 24 h. Subsequently, a scratch was gently made using a sterile pipette tip, and the cells were then stimulated with 20 ng/mL PDGF-ββ with or without atorvastatin calcium. The wounds were visualized and photographed at 0 h (immediately) and 24 h after the scratch.
Modified boyden chamber assay
Cell migration was also performed using a modified Boyden chamber model (Transwell, 8.0 m pore size, Corning, NY) as previously described. Cells suspensions were seeded in the upper chambers. PDGF-ββ with or without ATV was added to the bottom chamber as the chemoattractant. The cells were allowed to migrate through the membrane to the lower surface for 24 h. Cells on the upper surface of the membrane that had not migrated were scraped off with cotton swabs, and cells that had migrated to the lower surface were fixed by 3.7% paraformaldehyde, stained with DAPI, and counted. The migrated cell numbers were calculated as the number of migrated cells per 5 different random high-power fields.
Western blot analysis
The cell lysates were separated using sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE; 10-15% acrylamide gels) and then transferred to nitrocellulose membranes (Millipore, MA, USA) using a standard SDS-PAGE gel electrophoresis procedure. Membranes were then blocked at room temperature in Tris-buffered saline containing 0.1% Tween 20 (TBST) and 5% skim milk powder for 1 h. Membranes were incubated with primary antibodies for 4 h, and horseradish peroxidase (HRP)-conjugated secondary antibodies were used for detection. Primary antibodies used in this study were the following: anti-PCNA, anti-CDK2, anti-CDK4, anti-cyclin D1, anti-cyclin E, anti-phospho-PDGF-Rβ, anti-PDGF-Rβ, anti-phospho-Akt (Ser473), anti-Akt, anti-phospho-PLC-γ1, anti-PLC-γ1, anti-phospho-ERK1/2, anti-ERK1/2, and anti-GAPDH. Signal intensity was quantified using ImageJ 1.47 software (NIH, USA).
Zymography
MMP-2 and MMP-9 activities in the medium were analyzed by non-reducing SDS-PAGE in 10% gels containing 0.1% gelatin as previously described [27]. The zymograms were photographed and proteolysis was detected as a white zone in a blue background.
Statistical analysis
All variables were tested in three independent cultures for each experiment. Data are reported as the mean ± standard deviation (SD) and were analyzed using one-way analysis of variance (ANOVA). All analyses were performed using SPSS 18.0 statistical software (SPSS, Inc, Chicago, IL, USA). P<0.05 was considered to indicate a statistically significant difference.
Results
Inhibitory effect of atorvastatin calcium on PDGF-ββ-treated proliferation of VSMCs
We first examined the effect of atorvastatin calcium on PDGF-ββ-treated proliferation of VSMCs by direct cell counting, MTT assay and EdU incorporation assay. Stimulation with PDGF-ββ (20 ng/ml) for 24 h resulted in a significant increase in the number of VSMCs, which was decreased by atorvastatin calcium pretreatment in a concentration-dependent manner (Fig. 1A). In addition, atorvastatin calcium treatment in the absence of PDGF-ββ did not decrease the proliferation of VSMCs compared with the blank control (p>0.05). Similarly, the MTT assay showed the same inhibitory pattern on VSMC proliferation (Fig. 1B). Cell proliferation was further confirmed by [3H]-thymidine DNA incorporation. As shown in Fig. 1C and D, PDGF-ββ treatment significantly increased the DNA synthesis of VSMCs up to 1.7-fold compared with the untreated cells. DNA synthesis was inhibited by co-administration of PDGF-ββ and atorvastatin calcium (1, 5 and 10 µM). These results indicated that atorvastatin calcium played an inhibitory effect on PDGF-ββ-treated VSMCs proliferation.

Inhibitory effect of atorvastatin calcium on PDGF-ββ-treated proliferation of VSMCs. After 24h of starvation with serum-free DMEM, VSMCs were incubated with 20ng/mL PDGF-ββ and increasing concentrations (1, 5, 10µ M) of atorvastatin calcium (ATV) for 24h. (A) VSMCs were trypsinized, and their numbers were determined using hemocytometer. (B) VSMCs viability was evaluated by MTT assay. (C) EdU fluorescence staining to detect the newly synthesized DNA. (D) The percentage of EdU (+) cells was calculated by ImageJ 1.47 software. ∗P < 0.05 compared with 20ng/mL PDGF-ββ-treated controls.
Inhibitory effect of atorvastatin calcium on PDGF-ββ-treated proliferation of VSMCs. After 24h of starvation with serum-free DMEM, VSMCs were incubated with 20ng/mL PDGF-ββ and increasing concentrations (1, 5, 10µ M) of atorvastatin calcium (ATV) for 24h. (A) VSMCs were trypsinized, and their numbers were determined using hemocytometer. (B) VSMCs viability was evaluated by MTT assay. (C) EdU fluorescence staining to detect the newly synthesized DNA. (D) The percentage of EdU (+) cells was calculated by ImageJ 1.47 software. ∗P < 0.05 compared with 20ng/mL PDGF-ββ-treated controls.
The effect of atorvastatin calcium on PDGF-ββ-treated cell cycle progression
We determined whether atorvastatin calcium may modulate the cell cycle progression of PDGF-ββ-treated VSMCs using flow cytometry analysis. Serum deprivation of VSMCs for 24 h led to a 2.51% synchronization in the S phase of cell cycle progression. Compared with the control group, stimulation with 2 µM atorvastatin calcium alone did not affect the cell cycle progression. PDGF-ββ increased the percentage of cells into the S phase (from 2.51% to 28.88%) with a concomitant decrease in cells in the G0/G1 phase. However, 2 µM atorvastatin calcium in PDGF-ββ-treated VSMCs significantly reduced the percentage of cells in the S phase to approximately 16.82%, indicating that atorvastatin calcium may induce G0/G1 phase arrest in VSMCs (Fig. 2).
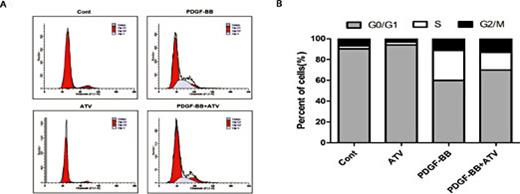
The effect of atorvastatin calcium on PDGF-ββ-treated cell cycle progression. After 24h of starvation with serum-free DMEM, VSMCs were incubated with 20ng/mL PDGF-ββ with or without ATV for 24h. (A) Individual nuclear DNA content was reflected by the fluorescence intensity of incorporated propidium iodide. Each item is derived from representative experiments, where data from at least 10,000 events were obtained. (B) The percentages of cell population at each stage are expressed as Mean values from three independent experiments.
The effect of atorvastatin calcium on PDGF-ββ-treated cell cycle progression. After 24h of starvation with serum-free DMEM, VSMCs were incubated with 20ng/mL PDGF-ββ with or without ATV for 24h. (A) Individual nuclear DNA content was reflected by the fluorescence intensity of incorporated propidium iodide. Each item is derived from representative experiments, where data from at least 10,000 events were obtained. (B) The percentages of cell population at each stage are expressed as Mean values from three independent experiments.
The effect of atorvastatin calcium on cell cycle regulatory protein expression
Eukaryotic cell growth is governed by cell cycle progression, and a growth signal activates an interaction between CDKs and cyclins. To investigate the mechanism of cell cycle arrest by atorvastatin calcium, we next analyzed the effect of atorvastatin calcium on the expression of cell cycle regulatory proteins, which could be involved in the G0/G1 arrest (Fig. 3). PDGF-ββ markedly increased in the cellular levels of PCNA, cyclin D1, CDK4, cyclin E and CDK2 in VSMCs (Fig. 3). In contrast, 2 µM atorvastatin calcium treatment attenuated the expression of PCNA, cyclin D1, CDK4, cyclin E and CDK2 in PDGF-ββ-stimulated VSMCs. These results suggested that atorvastatin calcium inhibited the cell cycle progression at the S phase via G0/G1 arrest.
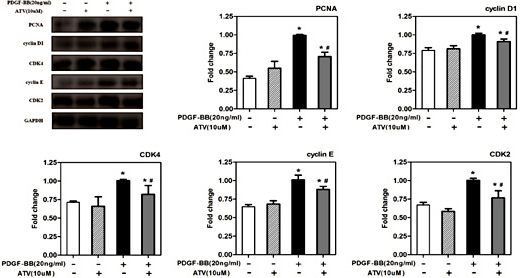
The effect of atorvastatin calcium on cell cycle regulatory protein expression. After 24h of starvation with serum-free DMEM, VSMCs were incubated with 20ng/mL PDGF-ββ with or without ATV for 24h. The protein expression levels of PCNA, cyclin D1, CDK4, cyclin E and CDK2 were determined by western blotting. GAPDH, glyceraldehyde 3-phosphate dehydrogenase. The band intensity was quantified using ImageJ 1.47 software and relative expression averaged across the three experiments. Probability values are indicated above bars: ∗P< 0.05 compared with nonstimulated controls; #P< 0.05 compared with 20ng/mL PDGF-BB-induced controls.
The effect of atorvastatin calcium on cell cycle regulatory protein expression. After 24h of starvation with serum-free DMEM, VSMCs were incubated with 20ng/mL PDGF-ββ with or without ATV for 24h. The protein expression levels of PCNA, cyclin D1, CDK4, cyclin E and CDK2 were determined by western blotting. GAPDH, glyceraldehyde 3-phosphate dehydrogenase. The band intensity was quantified using ImageJ 1.47 software and relative expression averaged across the three experiments. Probability values are indicated above bars: ∗P< 0.05 compared with nonstimulated controls; #P< 0.05 compared with 20ng/mL PDGF-BB-induced controls.
Inhibitory effects of atorvastatin calcium on PDGF-ββ-treated VSMCs migration
To evaluate the effect of atorvastatin calcium on the PDGF-ββ-treated migration of VSMCs, we performed two different migration assays. As shown in Fig. 4A, a wound-healing assay was performed. Treatment with PDGF-ββ for 24 h narrowed the wound distance and increased migration of VSMCs by about 5-fold after 24 h of treatment. Compared with PDGF-ββ treatment alone, atorvastatin calcium pretreatment attenuated PDGF-ββ-treated migration of VSMCs to approximately 70%. Next, we examined the effect of atorvastatin calcium on PDGF-ββ-treated VSMCs migration by Transwell assay. Like the results of wound-healing assay, we found the atorvastatin calcium could suppress the PDGF-ββ-treated VSMCs migration (Fig. 4B).

Inhibitory effects of atorvastatin calcium on PDGF-ββ-treated VSMCs migration. (A) Wound-healing assay showed that ATV inhibited PDGF-ββ-treated VSMCs migration, 100×. (B) Transwell assay was used for the evaluation of the migration of VSMCs. Magnification, 100×. Cell migration was determined by counting the cells that migrated through the pores. The results were presented as the means ± SEM from 3 different experiments. ∗P< 0.05 compared with nonstimulated controls; #P< 0.05 compared with 20ng/mL PDGF-ββ-treated controls.
Inhibitory effects of atorvastatin calcium on PDGF-ββ-treated VSMCs migration. (A) Wound-healing assay showed that ATV inhibited PDGF-ββ-treated VSMCs migration, 100×. (B) Transwell assay was used for the evaluation of the migration of VSMCs. Magnification, 100×. Cell migration was determined by counting the cells that migrated through the pores. The results were presented as the means ± SEM from 3 different experiments. ∗P< 0.05 compared with nonstimulated controls; #P< 0.05 compared with 20ng/mL PDGF-ββ-treated controls.
Atorvastatin calcium inhibits migration-related protein expression and VSMCs migration induced by PDGF- ββ
Next, we studied the underlying mechanisms of atorvastatin calcium on PDGF-ββ-treated VSMC migration through Western blot analysis (Fig. 5). It is reported that matrix metalloproteinases (MMPs) are involved in extracellular matrix (ECM) breakdown and are related to the development of atherosclerosis [28]. Thus, we examined the effect of atorvastatin calcium on several migration-regulated proteins and adhesion molecules, such as intercellular adhesion molecule 1 (ICAM-1), vascular cell adhesion molecule 1 (VCAM-1), MMP-2, and MMP-9. Our results revealed that atorvastatin calcium pretreatment inhibited the upregulation of ICAM-1, VCAM-1, MMP-2, and MMP-9 induced by PDGF-ββ. Furthermore, the zymography assay showed that atorvastatin calcium decreased MMP-2 and MMP-9 activities induced by PDGF-ββ in VSMCs. These findings suggested that atorvastatin calcium pretreatment reduced PDGF-ββ-treated VSMCs migration by suppressing the expression of migration-related proteins.
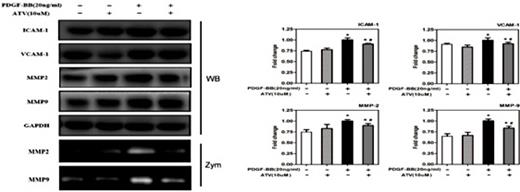
Atorvastatin calcium inhibits migration-related proteins expression and VSMCs migration induced by PDGF-ββ. After 24h of starvation with serum-free DMEM, VSMCs were incubated with 20ng/mL PDGF-ββ with or without ATV for 24h. The protein expression levels of ICAM-1, VCAM-1, MMP-2, and MMP-9 were determined by western blotting, and the enzymatic activities of MMP-2 and MMP-9 in the conditioned medium were measured by gelatin zymography assay. The band intensity was quantified using ImageJ 1.47 software and relative expression averaged across the three experiments. *P< 0.05 compared with nonstimulated controls; #P< 0.05 compared with 20ng/mL PDGF-ββ-treated controls. WB, Western blot; Zym, Zymograph.
Atorvastatin calcium inhibits migration-related proteins expression and VSMCs migration induced by PDGF-ββ. After 24h of starvation with serum-free DMEM, VSMCs were incubated with 20ng/mL PDGF-ββ with or without ATV for 24h. The protein expression levels of ICAM-1, VCAM-1, MMP-2, and MMP-9 were determined by western blotting, and the enzymatic activities of MMP-2 and MMP-9 in the conditioned medium were measured by gelatin zymography assay. The band intensity was quantified using ImageJ 1.47 software and relative expression averaged across the three experiments. *P< 0.05 compared with nonstimulated controls; #P< 0.05 compared with 20ng/mL PDGF-ββ-treated controls. WB, Western blot; Zym, Zymograph.
Atorvastatin calcium inhibits the activation of PDGF-Rβ signaling and Akt signaling pathways
PDGF-ββ binding to the PDGF receptor leads to the activation of several intracellular signaling cascades [29-31]. We next determined whether atorvastatin calcium affects the activation of PDGF-Rβ and its downstream signaling molecules, such as (PI3K)/Akt, (PLC)-γ1 and (ERK) 1/2 (Fig. 6). Treatment with PDGF-ββ for 5 min markedly increased the phosphorylation of PDGFRβ, (PLC)- γ1 and (ERK) 1/2 in VSMCs. Moreover, the phosphorylation of Akt was significantly increased after PDGF-ββ treatment for 15 min in VSMCs. In addition, atorvastatin calcium significantly inhibited PDGF-ββ-treated PDGFRβ phosphorylation (Fig. 6A) and Akt phosphorylation (Fig. 6B) in VSMCs. However, atorvastatin calcium treatment had no significant effect on the PDGF-ββ-treated phosphorylation of (PLC)-γ1 (Fig. 6C) or (ERK) 1/2 (Fig. 6D) in VSMCs. These results indicated that atorvastatin calcium may inhibit cell proliferation through the PDGFRβ and Akt signaling pathways.
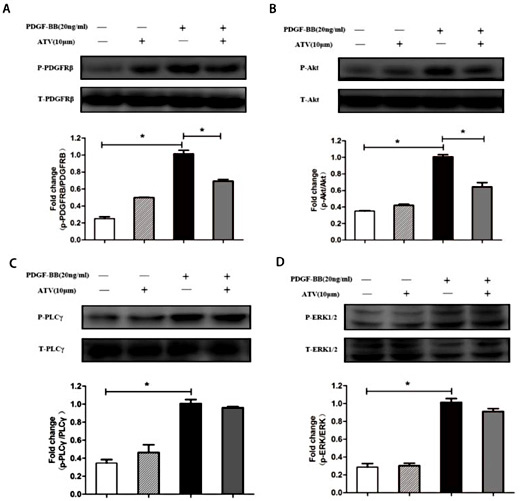
Atorvastatin calcium inhibits the activation of PDGF-Rβ signaling and Akt signaling pathways. VSMCs were pre-cultured in the presence or absence of ATV in serum-free medium for 30min, and then stimulated with 20ng/mL PDGF-ββ for 5min for the phosphorylation of PDGFRβ, (PLC)-γ1 and (ERK) 1/2, or 15min for Akt phosphorylation. Quantification of normalized densities for PDGFRβ, (PLC)-γ1, (ERK) 1/2 and Akt was shown. The graphs represent the relative activity of these kinases for three independent experiments. (A): PDGFRβ phosphorylation; (B): Akt phosphorylation; (C): (PLC)-γ1 phosphorylation; (D): (ERK) 1/2 phosphorylation. ∗P< 0.05 compared with nonstimulated controls; #P< 0.05 compared with 20ng/mL PDGF-ββ-treated controls.
Atorvastatin calcium inhibits the activation of PDGF-Rβ signaling and Akt signaling pathways. VSMCs were pre-cultured in the presence or absence of ATV in serum-free medium for 30min, and then stimulated with 20ng/mL PDGF-ββ for 5min for the phosphorylation of PDGFRβ, (PLC)-γ1 and (ERK) 1/2, or 15min for Akt phosphorylation. Quantification of normalized densities for PDGFRβ, (PLC)-γ1, (ERK) 1/2 and Akt was shown. The graphs represent the relative activity of these kinases for three independent experiments. (A): PDGFRβ phosphorylation; (B): Akt phosphorylation; (C): (PLC)-γ1 phosphorylation; (D): (ERK) 1/2 phosphorylation. ∗P< 0.05 compared with nonstimulated controls; #P< 0.05 compared with 20ng/mL PDGF-ββ-treated controls.
PDGFRβ inhibition attenuates the increased phosphorylation of Akt elicited by PDGF-ββ in VSMCs
Our data indicated that PDGF-ββ activated both the PDGFRβ and Akt signaling pathways. In addition, previous studies reported that there was cross-talk between the PDGFRβ and the (PI3K)/Akt pathways [13, 14]. Accordingly, AG1295 (a PDGFRβ-specific inhibitor) and LY294002 (an inhibitor of PI3K) were used in our study. As shown in Fig. 7, the inhibitors effectively inhibited the phosphorylation of PDGFRβ and Akt, respectively. Furthermore, the PDGFRβ-specific inhibitor (AG1295) not only abolished phosphorylation of PDGFRβ but also inhibited Akt activation. However, the PI3K/Akt inhibitor (LY294002) only decreased the phosphorylation of Akt; there was no detectable effect on PDGFRβ phosphorylation. These findings indicated that PDGFRβ likely served as an upstream regulator of Akt in PDGF-ββ-treated VSMCs.

PDGFRβ inhibition attenuates the increased phosphorylation of Akt elicited by PDGF-ββ in VSMCs. PDGFRβ-specific inhibitor (AG1295) abolished phosphorylation of both PDGFRβ and Akt, whereas PI3K/ Akt inhibitor (LY294002) had no detectable effect on PDGFRβ phosphorylation. The results were presented as the means ± SEM from 3 different experiments. ∗P< 0.05 compared with nonstimulated controls; #P< 0.05 compared with 20ng/mL PDGF-ββ-treated controls.
PDGFRβ inhibition attenuates the increased phosphorylation of Akt elicited by PDGF-ββ in VSMCs. PDGFRβ-specific inhibitor (AG1295) abolished phosphorylation of both PDGFRβ and Akt, whereas PI3K/ Akt inhibitor (LY294002) had no detectable effect on PDGFRβ phosphorylation. The results were presented as the means ± SEM from 3 different experiments. ∗P< 0.05 compared with nonstimulated controls; #P< 0.05 compared with 20ng/mL PDGF-ββ-treated controls.
The effect of atorvastatin calcium on PDGF-ββ-treated distribution of p-PDGFRβ and p-Akt in VSMCs
The above results revealed that atorvastatin calcium significantly inhibited PDGF-ββ-treated PDGFRβ phosphorylation and Akt phosphorylation in VSMCs. Then, we evaluated whether the distribution of p-PDGFRβ and p-Akt expression was affected by atorvastatin calcium in PDGF-ββ-treated VSMCs. As shown in Fig. 8A, compared with the control group, both the cytoplasmic and nuclear expression levels of p-PDGFRβ were significantly elevated in PDGF-ββ-treated VSMCs, and p-PDGFRβ expression was stronger in the cytoplasm than in the nucleus. Moreover, pretreatment of atorvastatin calcium resulted in p-PDGFRβ translocation into the nucleus. As shown in Fig. 8B, the expression of p-Akt was barely detectable in the control or atorvastatin calcium alone groups. In the PDGF-ββ-treated VSMCs, we found that strong fluorescent spots of p-Akt expression were gathered around the cell membranes. This result indicated that PDGF-ββ-treated Akt activation started by p-Akt accumulation around cell membrane, thus the phosphorylation of Akt might be carried out on the cell membrane, and then activated p-Akt was translocated to the cytoplasm and nucleus. We also found that pretreatment with atorvastatin calcium decreased the expression of p-Akt in PDGF-ββ-treated VSMCs, and fluorescent spots of p-Akt around the cell membrane disappeared (Fig. 8B).

The effect of atorvastatin calcium on PDGF-ββ-treated distribution of p-PDGFRβ and p-Akt in VSMCs. Immunofluorescence staining was performed to determine the expression and distribution of p-PDGFRβ (A) and p-Akt (B), 400×. The expression of p-PDGFRβ and p-Akt were red, and nuclei were stained blue with DAPI. The micrographs shown in this Fig. are representative of three independent experiments with similar results.
The effect of atorvastatin calcium on PDGF-ββ-treated distribution of p-PDGFRβ and p-Akt in VSMCs. Immunofluorescence staining was performed to determine the expression and distribution of p-PDGFRβ (A) and p-Akt (B), 400×. The expression of p-PDGFRβ and p-Akt were red, and nuclei were stained blue with DAPI. The micrographs shown in this Fig. are representative of three independent experiments with similar results.
Synergistic effects of atorvastatin calcium with Akt inhibitor on VSMCs proliferation
We determined whether the synergistic inhibition of VSMCs proliferation by atorvastatin calcium may occur with simultaneous treatment of Akt inhibitor. The expression of PCNA and Akt phosphorylation decreased in VSMCs treated with LY294002 (an Akt inhibitor) and atorvastatin calcium, compared to those of the compound alone-treated cells. These results indicated that the inhibitory effects of atorvastatin calcium on Akt phosphorylation were sufficient to suppress the PDGF-BB-stimulated proliferation of VSMCs (Fig. 9).

Synergistic effects of atorvastatin calcium with Akt inhibitor on proliferation of VSMCs and expression of PCNA and p-Akt. (A) After 24h of starvation with serum-free DMEM, VSMCs were incubated in the presence or absence of the indicated concentration of atorvastatin calcium or LY294002 (Akt inhibitor, 1uM) with 20ng/mL PDGF-ββ for 24h. VSMCs viability was evaluated by MTT assay. (B) VSMCs were pre-cultured in the presence or absence of ATV and LY294002 in serum-free medium for 30min, and then stimulated with 20ng/mL PDGF-ββ for 5min (for Akt phosphorylation) or 24h (for PCNA expression). Quantification of normalized densities for p-Akt and PCNA was shown in C&D. The graphs represent the relative activity of these kinases for three independent experiments. P< 0.05 compared with 20ng/mL PDGF-ββ-treated groups; #P< 0.05 compared with 20ng/mL PDGF-ββ+10uM ATV induced groups.
Synergistic effects of atorvastatin calcium with Akt inhibitor on proliferation of VSMCs and expression of PCNA and p-Akt. (A) After 24h of starvation with serum-free DMEM, VSMCs were incubated in the presence or absence of the indicated concentration of atorvastatin calcium or LY294002 (Akt inhibitor, 1uM) with 20ng/mL PDGF-ββ for 24h. VSMCs viability was evaluated by MTT assay. (B) VSMCs were pre-cultured in the presence or absence of ATV and LY294002 in serum-free medium for 30min, and then stimulated with 20ng/mL PDGF-ββ for 5min (for Akt phosphorylation) or 24h (for PCNA expression). Quantification of normalized densities for p-Akt and PCNA was shown in C&D. The graphs represent the relative activity of these kinases for three independent experiments. P< 0.05 compared with 20ng/mL PDGF-ββ-treated groups; #P< 0.05 compared with 20ng/mL PDGF-ββ+10uM ATV induced groups.
Discussion
In the present study, we investigated the anti-proliferative and anti-migratory activities of atorvastatin calcium on PDGF-ββ-treated VSMCs and the related signal transduction pathways. Proliferation of VSMCs plays an essential role in a broad spectrum of cardiovascular disorders [3]. Neointimal thickening is mainly due to the abnormal proliferation and migration of VSMCs from the vascular media. Thus, inhibition of VSMC proliferation and migration represents a potentially important therapeutic strategy for the treatment of cardiovascular disorders [2]. Among many known growth factors, PDGF-ββ is one of the most important mitogens and chemoattractants for VSMCs and plays a central role in proliferative vascular diseases. Atorvastatin calcium is a selective HMG-CoA reductase inhibitor that has pleiotropic biological effects, which include inhibiting HMG-CoA reductase activity, increasing LDL receptor levels and inhibiting VLDL-C synthesis. Increasingly, evidence suggests that statins have effects beyond reducing cholesterol levels. In our previous study, we demonstrated that atorvastatin calcium inhibited the phenotypic modulation of PDGF-ββ-treated VSMCs [26]. Furthermore, we conducted this study to investigate the related mechanisms under which atorvastatin calcium inhibited the abnormal proliferation and migration of PDGF-ββ-treated VSMCs.
Our study indicated that the anti-proliferative effect of atorvastatin calcium was associated with cell cycle arrest in the G0/G1 phase. Cell cycle progression is tightly regulated through a complex network of regulatory molecules, including cyclins and CDKs. CDKs promote the G-to-S phase transition by phosphorylation of the Rb protein to form a gene product-PCNA [32]. Cyclin D, cyclin E, CDK2, and CDK4 are known as positive mediators during the progression from the G0/G1 to the S phase of the cell cycle [33, 34]. A previous study showed that regulation of the cell cycle could inhibit VSMCs proliferation [35]. Thus, the cell cycle arrest was considered an effective strategy for the inhibition of VSMCs proliferation. Our data indicated that atorvastatin calcium permitted VSMCs arrest in the G0/G1 phase and inhibited the expression of the cell cycle regulatory proteins PCNA, cyclin D1, CDK4, cyclin E and CDK2 in PDGF-ββ-treated VSMCs compared to stimulation with PDGF-ββ alone. This result demonstrated that atorvastatin calcium inhibition of the PDGF-ββ-treated VSMC proliferation was due to the downregulation of the cycle regulatory protein expression, which resulted in the G0/G1 arrest.
During the process of pathogenic vascular disorders, dysfunctional VSMCs lead to abnormal migration activity [6]. Previous studies showed that MMPs, specifically MMP-2 and MMP-9, were activated rapidly following vascular injury [36-38] and played an important role in disrupting the ECM in VSMCs [37, 39]. Degradation of the ECM promoted VSMC migration from the media of arterial walls to the intimal space, where VSMCs secreted more ECM products, progressively resulting in restenosis and neointima formation [40, 41]. Moreover, several migration regulatory proteins, including cell adhesion molecules ICAM-1, VCAM-1 [27, 42], MMP2, and MMP-9 were reported to be vital regulators of abnormal VSMC migration [43-45]. In the present study, our results indicated that atorvastatin calcium inhibited the expression of ICAM-1, VCAM-1, MMP2, and MMP-9 induced by PDGF-ββ stimulation. This demonstrated that atorvastatin calcium inhibited PDGF-ββ-treated VSMC migration, at least partially owing to the suppression of the increased expression of migration regulatory proteins induced by PDGF-ββ.
In this study, we showed that PDGF-ββ induced PDGF-R phosphorylation in VSMCs, and PDGF-ββ activity was altered by atorvastatin calcium. PDGF-ββ also activated many downstream signaling molecules, such as (PI3K)/Akt, (PLC)-γ1 and (ERK)1/2 pathways, which are involved in cell growth and survival [46-48]. We found that phosphorylation of PDGFRβ, Akt, (PLC)-γ1 and (ERK) 1/2 was induced by PDGF-ββ; atorvastatin calcium inhibited PDGF-ββ-treated phosphorylation of Akt but not (PLC)-γl or (ERK) 1/2. As we found that both the PDGFRβ and (PI3K)/Akt pathways were activated following stimulation by PDGF-ββ and there was cross-talk between the PDGFRβ and (PI3K)/Akt pathways, we sought to determine whether Akt or PDGFRβ served as the upstream molecule in VSMCs stimulated by PDGF-ββ. We observed that blocking the PDGFRβ pathway with a specific inhibitor abolished the phosphorylation of both PDGFRβ and Akt, whereas PDGFRβ phosphorylation was not affected following Akt inhibition in VSMCs. Our results indicated that PDGFRβ served as an upstream regulator of Akt in the setting of PDGF-ββ treatment in VSMCs.
Conclusion
The present study indicated that atorvastatin calcium inhibited abnormal proliferation and migration of VSMCs through G0/G1 cell cycle arrest and suppression of PDGFRβ-PI3K-Akt signaling cascade.
Disclosure Statement
The authors declare that there is no Disclosure Statement.
Acknowledgements
This study was supported by Key Laboratory of Myocardial Ischemia, Harbin Medical University, Chinese Ministry of Education to Dr. Shuang Chen (No.KF201509).

