

Abstract
Objective: To use functional magnetic resonance imaging (fMRI) in craniopharyngioma (CP) patients to examine the hypothesis that hypothalamic damage due to CP and its treatment results in enhanced perception of food reward and/or impaired central satiety processing. Methods: Pre- and post-meal responses to visual food cues in brain regions of interest (ROI; bilateral nucleus accumbens, bilateral insula, and medial orbitofrontal cortex) were assessed in 4 CP patients versus 4 age- and weight-matched controls. Stimuli consisted of images of high- (‘fattening’) and low-calorie (‘non-fattening’) foods in blocks, alternating with non-food object blocks. After the first fMRI scan, subjects drank a high-calorie test meal to suppress appetite, then completed a second fMRI scan. Within each ROI, we calculated mean z-scores for activation by fattening as compared to non-fattening food images. Results: Following the test meal, controls showed suppression of activation by food cues while CP patients showed trends towards higher activation. Conclusion: These data, albeit in a small group of patients, support our hypothesis that perception of food cues may be altered in hypothalamic obesity (HO), especially after eating, i.e. in the satiated state. The fMRI approach is encouraging for performing future mechanistic studies of the brain response to food cues and satiety in patients with hypothalamic or other forms of childhood obesity.
Introduction
One of the most striking examples of childhood obesity is observed in children with craniopharyngioma (CP), an embryological tumor located in the hypothalamic and/or pituitary region. CPs are tumors that originate in ectodermic cell remains of the Rathke pouch and grow along midline structures. About 20% of CPs show exclusively suprasellar localization and 5% are intrasellar [1]. Tumor resection with and without radiotherapy is the therapeutic standard of care. According to published studies, partial tumor resection followed by radiation therapy leads to tumor control in more than 80% of cases [2,3,4,5]. After treatment, patients often suffer from partial hypopituitarism or panhypopituitarism. Despite optimal endocrine management, uncontrolled appetite and rapid weight gain are common, possibly resulting from destruction of parts of the medial hypothalamus, such as the ventromedial hypothalamus [6], while only few patients were reported suffering from anorexia [7,8,9].
Excessive weight gain is one of the most distressing manifestations of hypothalamic injury associated with CP. In a large longitudinal study on growth and BMI utilizing German reference data in 90 children who underwent surgery for CP, Muller et al. [10] found that reduced growth rates occurred early in the course of CP. In 30% of all patients with hypothalamic tumors, BMI is increased at the time of diagnosis before surgery (mean BMI standard deviation score (SDS) at diagnosis CP with hypothalamic involvement 1.1 vs. 0.3 in CP without hypothalamic involvement), although their BMI at birth was below average based on population-based reference data [10]. Of the 42 patients without hypothalamic involvement, 69 % maintained normal body weight (BMI SDS < 2), while only 10% of the 48% patients with hypothalamic involvement maintained normal-weight status. Neither the frequencies of irradiation (29% vs. 20%) nor the frequencies of endocrine disorders were significantly different in these two groups. These data clearly demonstrate that i) hypothalamic location of the tumor and ii) surgery in the hypothalamic area are risk factors for the development of obesity.
After surgery, hyperphagia and obesity occur on average in about 50% of all CP patients [11,12,13], although study results vary from 6% [14] to 91% [10,15], with higher rates in CP patients with hypothalamic damage compared to CP without hypothalamic damage. In children and adolescents, the incidence of severe obesity following CP resection surgery is between 22% and 62% [16,17,18]. Treatment of hyperphagia and hypothalamic obesity (HO) is extremely difficult. Attempts at behavior modification have also been largely unsuccessful [19]. Only a few reports have been published on treating HO with drugs [20,21,22], and, compared to uncomplicated obesity, weaker weight reductions were observed [21].
Risk factors for developing obesity in CP patients include large hypothalamic lesions affecting medial hypothalamic nuclei such as the arcuate nucleus (ARC), ventromedial nucleus (VMN), and the dorsomedial nucleus (DMN); tumors that reach the floor of the third ventricle; hydrocephalus; transcranial surgery, which often causes greater damage to the hypothalamus compared to transnasal surgical tumor removal; aggressiveness of resection; reoperation for tumor recurrence; and hypothalamic irradiation [23,24,25,26]. Among them extensive hypothalamic infiltration and a completely deficient third ventricle have been associated with the highest risk of obesity [27]; however, results are not entirely consistent [28]. Disturbance of the hypothalamus by the tumor itself and its surgical treatment have been discussed as major causes of hyperphagia and obesity [26]. VMN damage can lead to disinhibition of the vagal tone, resulting in excess stimulation of pancreatic β-cells, hyperinsulinemia, and obesity [29]. Alternatively, sympathetic nervous output might be reduced [30], leading to decreased physical activity [31]. Because hypothalamic damage can include the ARC, which is the main hypothalamic binding site for adipostatic hormones coming from the periphery, the hypothalamus might be unable to respond normally to insulin and leptin, the key regulators of energy homeostasis, or to gut hormones including ghrelin [32,33]. Alternatively, disinhibition of orexigenic peptide-secreting neurons in the lateral hypothalamus, caused by injury to medial hypothalamic nuclei, may drive excess weight gain [34,35,36]. In sum, brain regulation of appetite and body weight is disturbed in CP patients post treatment, but the mechanisms by which CP-derived brain lesions promote excess food intake are unknown.
Functional magnetic resonance imaging (fMRI) is a powerful tool for observing the human brain’s in vivo responses to stimuli. fMRI techniques measure changes in the blood oxygen level-dependent (BOLD) response to indirectly quantify neural activity. These techniques have been successful in documenting the response of brain areas such as the prefrontal cortex, orbital frontal cortex, amygdala, thalamus, hypothalamus, and hindbrain to photographs of food [37,38,39]. fMRI has been successfully applied to the study of obesity. Indeed, early neuroimaging studies have documented differences between obese and lean adults in neural responses to satiating meals [40,41,42], taste [43,44], and food cues [45,46,47,48], especially in regions that process rewarding aspects of food intake. Findings in obese adults show greater activation by high-calorie visual food cues, compared to lean controls, in the striatum, orbital frontal cortex, insula, and hippocampus [46,47] as well as in the nucleus accumbens, amygdala, cingulate cortex, prefrontal cortex, and ventral pallidum [46]. Two published fMRI studies in obese children showed hyperresponsivity to food cues both before and after a meal in the prefrontal and orbitofrontal cortex [49,50].
Functional neuroimaging data in patients with CP is lacking and might aid in discriminating among the different theories for the development of obesity (i.e. size and location of tumor vs. post-surgical lesion vs. effect of irradiation). An important unanswered question is whether damage from CP alters function in brain regions involved in reward and/or satiety processing, especially since new therapies under development for obesity preferentially affect one or the other of these responses.
We describe data using fMRI that supports our hypothesis regarding the central mechanisms that drive hyperphagia and obesity in children with CP after tumor resection. By using a disease model in which central effects of major regulators of energy homeostasis, such as leptin and insulin, are disturbed [29,51,52], we postulate disturbed interactions between peripherally circulating peptides and neural centers that drive perceptions of hunger and satiety in patients with hypothalamic damage.
Material and Methods
Study Participants
We enrolled eight 13- to 17-year-old individuals, four with CP (2 male, 2 female, BMI 25.3–43.8 kg/m2) and four controls (1 male, 3 female, BMI 20.2–47.7 kg/m2). The study was performed at the University of Washington (UW) Medical Center Clinical Research Center. All procedures were approved by the UW Children’s Hospital institutional review board. Two normal-weight and 2 obese CP patients as well as 2 obese otherwise metabolically healthy controls without underlying endocrine disorders were recruited from the Seattle Children’s Hospital endocrine outpatient clinics. Two lean controls were recruited by announcement at Seattle Children’s Hospital. None of the 4 controls took any medications. CP patients were at least 1 year post surgery and post irradiation. All CP patients had risk factors for obesity as all had hypothalamic tumor location and had undergone frontal craniotomy. In 3 of the patients tumor excision was complete. One patient had disturbed cerebrospinal fluid circulation with a residual cyst after surgery which was treated by stereotaxic cyst aspiration, drainage, and intracavitary bleomycin instillation. Two CP patients received cranial irradiation. Two patients had panhypopituitarism including diabetes insipidus, and 2 patients partial hypopituitarism. Depending on their pituitary hormone deficiencies, CP patients were substituted with L-thyroxine, growth hormone, desmopressin, hydrocortisone, and sex steroids.
Study Procedures
After an overnight fast, study participants ate a small breakfast consisting of 10% of their estimated daily caloric requirements as calculated by the Mifflin St. Jeor formula [53]. The calculated energy of the standardized breakfast was 271 kcal for the CP group and 274 kcal for the control group. This controlled breakfast matched the groups for caloric intake prior to the first fMRI scan. Body composition was measured by dual energy X-ray absorptiometry (DEXA) scan in all subjects but 1 CP patient, whose body weight exceeded equipment limits. The first fMRI scan took place 3 h after the controlled meal. Immediately following the first fMRI scan, participants exited the MR scanner to drink a high-calorie test meal (milkshake) providing 20% of estimated daily calorie requirements and containing 55% of energy as carbohydrates, 15% as protein, and 30% as fat. The calculated energy of the milkshake was 542 kcal for the CP group and 548 kcal for the control group. The milkshake was given to suppress appetite before subjects were scanned ∼30 min later, a time period that is necessary to allow changes of secreted peptides that are involved in the regulation of food intake [54,55,56] in addition to a portion of the satiety response that is mechanical and likely vagally mediated [57]. Participants then went back into the scanner for the second fMRI scan. During the fMRI scans, participants viewed photographs of food and non-food objects (see fMRI stimuli below). Visual analogue scale (VAS) appetite questionnaires were conducted before and after each scan [58]. Subjects rated their current hunger and fullness by marking their rating on a line of 100 mm, anchored at each end by statements describing the extremes (e.g., ‘I am not hungry at all,’ ‘I have never been more hungry,’ or ‘Not at all full,’ ‘Totally full’) [58]. 1 h after the milkshake, an ad libitum buffet meal was served consisting of a wide variety of food items and more than 100% of the subject’s estimated daily calorie requirements. The lunch buffet was provided to objectively assess satiety. The meal had to be finished within 30 min. Subjects were not told that we will assess their calorie intake or composition of consumed foods after they finished the meal.
BOLD responses were measured by fMRI while adolescents viewed photographs grouped into ‘fattening’ food, ‘non-fattening’ food, and non-food objects. Stimuli were classified originally by independent adult raters as to whether or not they should be eaten when dieting. Food photographs in the fattening group were characterized by high-calorie content and were usually high in fat and/or sugar (fig. 1). Non-fattening food photographs depicted low-calorie foods, including fruits, vegetables, and low-fat meats. Non-food images consisted of common, recognizable large and small objects such as furniture, toiletries, sundries, electronics, and household items. Thus, the terminology ‘fattening’ encompasses both the social perception of the foods as well as their caloric content and energy density. We validated these photographs using questionnaires and interviews in young adults [39], and recently also in sixteen 8- to 16-year-old children. 91% of images were correctly classified as fattening (makes people gain too much weight) or non-fattening. As in adults, children rated the fattening and non-fattening foods as similarly appealing. The fMRI paradigm consists of 3 blocks of photographs from the fattening food group, 3 blocks of photographs from the non-fattening food group, and 7 blocks of non-food object photographs. All food and object images were commercial-quality stock photographs obtained from websites or donated for research use (Great American Stock, Brookfield, WI, USA).
Sample photographs. Example of pictures showing low-calorie a ‘non-fattening’ and b high-calorie ‘fattening’ food as well as c non-food items.
Sample photographs. Example of pictures showing low-calorie a ‘non-fattening’ and b high-calorie ‘fattening’ food as well as c non-food items.
Image Acquisition
Structural and fMRI exams were performed on a 3T Philips Achieva MR System (version 1.5, Philips Medical Systems, Best, the Netherlands) with dual Quasar gradients (80 mT/m with a slew rate of 110 mT/m/s or 40 mT/m at a slew rate of 220 mT/m/s) using an 8-channel SENSE head coil. Functional whole-brain T2*-weighted images were acquired by using a single-shot gradient-recalled echo-planer imaging (EPI) sequence (TR = 2,000 ms; TE = 30 ms; flip angle = 76°; FOV = 240 mm) with a matrix size of 64 × 64 (reconstructed in-plane resolution = 3 × 3 mm) (for details see Schur et al. [39]). This scanning protocol was used for both the pre- and post-milkshake session. The SameScan software program, part of the Philips scanning software platform (Version R2.5.3.0), was used to reposition the participants reproducibly for the 2 scans.
fMRI Paradigm and Processing
The fMRI paradigm consisted of 3 blocks of photographs from the high-calorie food group, 3 blocks of photographs from the low-calorie food group, and 7 blocks of non-food object photographs. Ten photographs were presented per block. Each photograph was projected for 2.4 s onto a screen easily viewed in a mirror by the participant. Non-food object blocks alternated with high-calorie and low-calorie food blocks. To ensure that subjects attended to the images, participants were advised before and during the fMRI scan to focus carefully on each image and were told that they would be tested later on which photographs they saw. In a behavioral post-test following each scan, subjects were asked to distinguish between 16 images seen in the scanner and 16 distractor images and the percent of correct responses was calculated. fMRI data analyses were performed using FSL (www.fmrib.ox.ac.uk/fsl/), similar to the study published by Schur et al. [39]. Condition effects were estimated at each voxel for the contrast of fattening food > non-fattening food. Individual fMRI data were registered to the high-resolution scan by utilizing fieldmap corrections, warped with an affine transformation to the MNI152 standard image using FMRIB’s Linear Image Registration Tool, and resampled to 2 mm3 voxels.
A priori ROIs have been previously shown to be differentially responsive to food photographs based on the energy content of the foods depicted. They include the striatum, nucleus accumbens, hypothalamus, orbital frontal cortex, brainstem, amygdala, insular cortex, and prefrontal cortex [39]. In this study, we focused on analyzing z-scores in five brain regions: bilateral nucleus accumbens, bilateral insula, and medial orbitofrontal cortex. ROIs were defined by Automatic Anatomical Labeling [59], except for the nucleus accumbens ROI which was defined by the Harvard-Oxford Subcortical Structural Atlas. Mean ROI z-scores for activation by fattening > non-fattening food photographs were extracted for each subject from both pre- and post-test meal scans.
Statistical Analysis
Normally distributed variables in groups were compared by Student’s t test for unpaired observations. Non-Gaussian variables were be compared by Mann-Whitney U test. In multiple group comparisons, data were analyzed by one-way analysis of variance (ANOVA) for multiple groups, followed by Bonferroni’s Multiple Comparison Test. Pair-wise two-group t-tests were employed post hoc as indicated.
Results
Investigation of body composition in 3 CP patients and 4 control subjects showed a tendency toward higher body fat and lower lean mass in CP subjects than controls (mean percentage body fat CP = 45.6 ± 3.0% vs. controls = 40.6 ± 5.0%; mean percentage lean mass CP = 53.0 ± 2.8% vs. controls = 57.2 ± 4.6%), although their mean BMI z-scores were comparable (CP = 1.43 ± 0.2 vs. controls = 1.42 ± 0.72).
Assessment of hunger and fullness by VAS showed that CP patients reported less hunger before the test meal (milkshake), and showed a trend toward less reduction of hunger by the meal (fig. 2a). They also reported less increase in subjective fullness after the test meal as compared to controls (fig. 2b). In addition, CP patients tended to have higher caloric intake at the ad libitum buffet lunch (fig. 2c), with higher intakes of fat (CP 59.9 ±19.5 g vs. controls 37.9 ±15.4 g) and sugar (CP 67.2 ±18.3 g vs. controls 53.7 ±23.2 g). However, given the small sample size, these trends were not statistically significant.
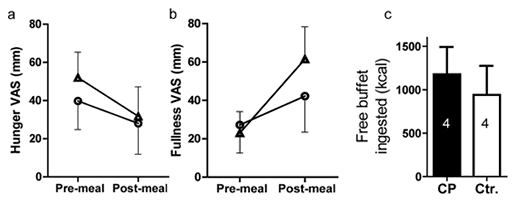
Subjective hunger, fullness and actual caloric intake in CP patients versus controls. Ratings of a hunger and b fullness from VAS before and 30 min after the test meal in the CP (circles) and control (triangles) groups are shown as mean values (± SEM). Possible scores ranged from 0–100 mm. Higher scores indicated more hunger or fullness. Panel c shows group means (± SEM) kilocalories ingested at an ad libitum buffet meal that immediately followed the second fMRI scan, occurring 60 min after the test meal. CP = Craniopharyngioma; Ctr = control.
Subjective hunger, fullness and actual caloric intake in CP patients versus controls. Ratings of a hunger and b fullness from VAS before and 30 min after the test meal in the CP (circles) and control (triangles) groups are shown as mean values (± SEM). Possible scores ranged from 0–100 mm. Higher scores indicated more hunger or fullness. Panel c shows group means (± SEM) kilocalories ingested at an ad libitum buffet meal that immediately followed the second fMRI scan, occurring 60 min after the test meal. CP = Craniopharyngioma; Ctr = control.
Before the test meal, CP patients tended to have lower BOLD activity in the five selected brain areas. 30 min after the test meal, controls showed a suppression of BOLD activity while CP patients showed a trend towards higher BOLD activity in these brain areas (fig. 3, fig. 4). Trends were strongest in the medial orbitofrontal cortex (MOFC) for the baseline study (p = 0.05; one-sided) as well as for the change in z-score from before to after the test meal (p = 0.04; one-sided).

Representative images from a control subject and a CP patient showing brain activation by visual food cues before and after a meal in the bilateral nucleus accumbens. Coronal sections through the nucleus accumbens in standard space are shown for 2 individual subjects at 2 time points, selected as representative of the mean z-score for the contrast of high-calorie > low-calorie food for each group. Control subject a before and b after the test meal. CP patient c before and d after the test meal. The decrease in voxel z-scores (change from yellow/orange to red/blue color) indicates suppression of activation by high-calorie food cues after a meal in the control subject but not in the CP patient. R = Right; L = left.
Representative images from a control subject and a CP patient showing brain activation by visual food cues before and after a meal in the bilateral nucleus accumbens. Coronal sections through the nucleus accumbens in standard space are shown for 2 individual subjects at 2 time points, selected as representative of the mean z-score for the contrast of high-calorie > low-calorie food for each group. Control subject a before and b after the test meal. CP patient c before and d after the test meal. The decrease in voxel z-scores (change from yellow/orange to red/blue color) indicates suppression of activation by high-calorie food cues after a meal in the control subject but not in the CP patient. R = Right; L = left.

Activation by food cues in CP patients vs. controls. a Mean activation in selected brain regions of interest (ROIs) from CP patients (black bars) and control subjects (white bars) before the test meal. b Change in activation from pre- to post-meal. Group means (± SEM) were calculated from individual mean z-scores for the contrast of high-calorie > low-calorie food extracted from each ROI. R = Right; L = left; Acc = nucleus accumbens; Ins = insula; MOFC = medial orbitofrontal cortex. *p = 0.05 versus control, #p = 0.04 versus control. P-values determined by one-sided Student’s t-tests.
Activation by food cues in CP patients vs. controls. a Mean activation in selected brain regions of interest (ROIs) from CP patients (black bars) and control subjects (white bars) before the test meal. b Change in activation from pre- to post-meal. Group means (± SEM) were calculated from individual mean z-scores for the contrast of high-calorie > low-calorie food extracted from each ROI. R = Right; L = left; Acc = nucleus accumbens; Ins = insula; MOFC = medial orbitofrontal cortex. *p = 0.05 versus control, #p = 0.04 versus control. P-values determined by one-sided Student’s t-tests.
Discussion
Several factors leading to HO in CP patients have been identified, but important unanswered questions remain, such as whether damage to homeostatic centers also alters reward-mediated eating behavior and whether central processing of satiety is affected. Eating behavior is critically regulated by hypothalamic nuclei as well as by non-homeostatic processes (including cognition, emotion, and reward processing) that are directed primarily by corticolimbic and higher cortical brain regions. We hypothesized that hypothalamic damage due to CP and/or its treatment results in enhanced perception of food reward and focused on brain regions regulating rewarding aspects of eating behavior. Following the test meal, controls showed suppression of activation by images of high-calorie, energy-dense food while CP patients showed trends towards higher activation in ROIs including the insula, nucleus accumbens, and MOFC. These results point to altered central satiety response within selected subcortical and cortical regions regulating rewarding aspects of food intake. Albeit in a small group of patients, these empiric data provide the first in vivo observations of responses to food cues in patients with CP, both before and after food intake. The findings support our hypothesis that perception of food cues may be altered in HO, especially after eating.
Further supportive evidence is demonstrated by a trend showing that CP patients also reported less change in subjective appetite after standardized test meals. Specifically, fullness ratings decreased less in CP patients than controls. Though these trends were statistically not significant, the findings are in line with the lack of suppression of BOLD activity in CP versus control subjects following the test meal.
According to our results, we postulate that medial hypothalamic damage may disrupt communication between the hypothalamus and corticolimbic pathways governing food reward, potentially exacerbating known disturbances in signaling by peripheral adiposity and satiety peptides. Our results support a model in which destruction of hypothalamic structures affects not only neurochemical communication within the hypothalamus but also communication with areas involved in reward processing (e.g., the nucleus accumbens) and behavior modification (e.g., the orbital frontal cortex).
Although central processing of the reward value of food has been implicated in simple adult obesity [43,46,47], multiple sites of action or altered function in children with HO could underlie our findings. fMRI studies using visual stimuli provide evidence that the same neural and hormonal mechanisms that regulate energy homeostasis and eating behavior may also modulate attention to, and perhaps perception of, environmental food cues. Leptin replacement in genetically deficient individuals suppresses activity stimulated by visual food cues in the nucleus accumbens [60] while heightening activity in prefrontal cortex regions has been linked to inhibition of food intake [61]. Leptin also reverses weight loss-induced changes in neural responses to food cues [62]. In contrast, ghrelin, an orexigenic hormone that shows high preprandial but low postprandial plasma levels [63] stimulates activity during viewing of food pictures in the amygdala, orbital frontal cortex, anterior insula, and striatum [64]. fMRI of normal-weight humans infused with PYY3–36, a gut hormone that is physiologically released in response to a meal, to circulating concentrations similar to those observed postprandially showed modulated neuronal activity within the hypothalamus, brainstem, and mid-brain regions involved in food reward processing [54]. This suggests that PYY3–36 acts on both homeostatic and hedonic brain circuits. There is good evidence for alterations in peripheral signaling by these hormones in CP. Leptin resistance has been found in CP patients with hypothalamic lesions [51]. In addition, patients with hypothalamic CP have weaker post-meal reductions in ghrelin concentrations than CP without hypothalamic tumors [25]. In addition, obese CP subjects have significantly higher baseline and post-meal insulin along with weaker post-meal changes in PYY [25]. In sum, prior studies of HO have shown impaired function of peripherally released hormones that act centrally to modulate responses to food cues. Thus, our findings could be the result of either disruption in peripheral signaling or in hypothalamic neural circuits communicating with corticolimbic and other brain regions to regulate appetite. For either deficit, our findings illustrate how alterations in brain function after CP might result in a failure to adjust perceptions of environmental food cues to the current state of energy needs, resulting in hyperphagia and weight gain.
The fMRI approach described is promising for investigating HO. The presented data supports our hypothesis that the post-meal suppression of neuronal activity induced by visual food cues in food reward brain areas is blunted in CP patients. However, additional imaging data is required to either support or refute our hypothesis as we only tested 4 subjects per group. We are aware that our small pilot study cannot discern effects caused by the tumor itself versus the surgery or irradiation because all of our CP patients were examined after surgery and 2 of them had also irradiation. As stated in the introduction, a large lesion affecting medial hypothalamic nuclei caused by extensive surgical removal of a large hypothalamic tumor might be the strongest risk factor for HO [11,65]. Future studies should include correlation analyses between fMRI changes and changes of specific endocrine regulators of food intake such as ghrelin and other gut hormones. A larger multicenter study would potentially allow performing subgroup analyses stratified for size and location of different hypothalamic lesions as well as different treatment modalities, such as frontal craniectomy versus transsphenoidal resection. In addition, longitudinal fMRI studies could be performed in CP patients before and directly after surgical or irradiation interventions. Furthermore, in addition to providing mechanistic insights, future fMRI studies in CP patients could inform more effective approaches for preventing HO in children with CP as well as testing pharmacological interventions.
Acknowledgments
The authors thank Dr. Thomas Grabowski of the University of Washington Integrated Brain Imaging Center and Dr. Kenneth Maravilla of the University of Washington Diagnostic and Imaging Sciences Center for constructive ideas and advice while developing the study protocol and interpreting the data. We also thank Susan Kearns, Seattle Children’s Hospital, for subject enrollment and data collection. This project was supported by a pilot study grant from the Center for Clinical and Translational Research, Seattle Children’s Hospital (PI-Roth). Dr. Schur’s time was supported by NIH K23 DK070826 (PI-Schur).
Disclosure Statement
The authors did not provide a conflict of interest statement.
