

Abstract
Background/Aims : Arterial hypertension is characterized by vascular remodelling, atherosclerosis and cardiovascular complications. Matrix metalloproteases (MPPs) are endopeptidases produced by all the cells present in the vascular wall and are involved in the regulation of the extracellular matrix protein turnover. MMPs contribute to blood vessel formation, remodelling, angiogenesis; whereas an altered expression or activity of MMPs or their tissue inhibitors (TIMPs) results correlated with the development and progression of cardiovascular complications. Methods: We examined the literature data regarding the role of MMPs in human hypertension, including their involvement in vascular remodelling, and the effects of some antihypertensive molecules on these MMP/TIMP profile. Results: The expression and the activity of some MMPs and TIMPs are impaired in human hypertension. An altered MMPs/TIMPs balance plays an important role in the vascular wall rearrangement, in response to hemodynamic changes which may induce myocardial hypertrophy and fibrosis leading to ventricular remodelling. Several studies have examined the effects of some antihypertensive molecules, such as ACE inhibitors, angiotensin receptor blockers, calcium-channel blockers, and aldosterone antagonists, on the MMPs/TIMPs profile by obtaining positive results. Conclusion: Considering the data taken into consideration, the authors believe that in clinical practice a strategic antihypertensive therapy directed to the MMPs profile, may be useful to decrease the risk of cardiovascular complications.
Introduction
Arterial hypertension is associated with vascular remodelling characterized by a rearrangement of vascular wall components, including extracellular matrix (ECM) protein, such as elastin and collagen fibres. Reciprocal interaction between vascular cells and their ECM is pivotal in blood vessel formation and remodelling [1]. Hypertensive vascular remodelling is an adaptive response to changes in the blood-pressure-induced circumferential wall stress and blood-flow-induced wall shear stress, which result in the degradation and reorganization of the vascular ECM protein, such as collagen, elastin, proteoglycan and fibronectin [2, 3].
Matrix metalloproteinases (MMPs) are a large variety of endopeptidases involved in ECM protein degradation by cleavage of internal peptide bonds. Each MMP has a specific substrate target that defines its denomination, such as collagenase, gelatinase, stromelysin and matrilysin.
Collagenases (MMP-1, -8, -13, and -18) cleave interstitial collagen I, II, and III, and also other molecules, such as bradykinin and angiotensin I. Fragments of collagen are then degraded by gelatinases [4]: MMP-2 and -9 are secreted by several vascular cell types, including endothelial cells, pericytes and podocytes, fibroblasts and myofibroblasts, monocyte derived macrophages and local macrophages tissue [5]. They are responsible for IV type collagen degradation, vasculature remodelling, angiogenesis, inflammation and atherosclerotic plaque rupture [6, 7]. MMP-2 is constitutively expressed on cell surface, while MMP-9 is stored in secretory granules in different cell types and it is inducible by exogenous stimuli [5, 8]. Stromelysin-1 (MMP-3) and -2 (MMP-10) degrade the fibronectin, laminin, gelatins-I, III, IV and V, collagen fibers, and proteoglycans [6]. Matrilysins may hydrolize fibronectin, gelatins [6] and may cleave plasminogen producing a fragment that inhibits angiogenesis. Membrane-Type MMPs (MT-MMPs), transmembrane or GPI (glycosylphosphatidylinositol)-anchored, can degrade type-I, -II, and III collagen and other components of ECM [4]. The regulation of MMPs synthesis and activity is highly complex. Radical oxygen species (ROS), growth factors, cytokines and hormones can influence MMP transcription through the activation of the Mitogen-Activated Protein Kinase (MAPK), the inhibition of MAPK phosphatase, the inactivation of the histone deacetylase (involved in gene repression) or the recruitment of different chromatin remodelling factors [9].
Most of the MMPs are synthesized as precursors (pro-MMP) and must be activated to expose the catalytic domain with the Zn2+–binding site. This activation can be effected by proteolytic modifications from several proteases [8, 10] and also by S-glutathiolation, S-nitrosylation and phosphorylation reactions [11, 12] (Fig. 1).
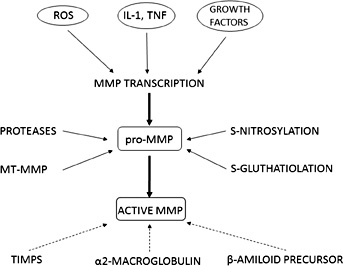
In experimental models, some authors have demonstrated that angiotensin II could promote the expression of MMP-8 and MMP-13 by macrophages and their activity in atherosclerotic plaques [13], while others have highlighted that MMP-2 mediates angiotensin II-induced hypertension, being transcriptionally regulated by MMP-7 and by a disintegrin and metalloproteases (ADAM-17) [14]. On the contrary, MMPs activity can be underegulated by several proteins, such as alpha2-macroglobulin, β-amiloid precursor protein and GPI-anchored glycoprotein, and by the four tissue inhibitors of MMP (TIMPs): TIMP-1 acts on MMP-1, MMP-3, MMP-7 and MMP-9; TIMP-2 inhibits especially MMP-2; TIMP-3 can inhibit MMP-2 and MMP-9; TIMP-4 inhibits MT-1 MMP and MMP-2 activity [8, 15] (Fig. 1). TIMPs activities include regulation of cell proliferation, migration and invasion, anti-angiogenesis and apoptosis [16]. Most of these activities arise from MMP inhibition, but TIMPs are also able to interact with some specific cell receptors; for example, TIMP-3 binds vascular endothelial growth factor (VEGF) receptor on endothelial cells inhibiting angiogenesis [16].
MMPs contribute to the remodelling of basement membranes and the degradation of the components of the ECM and are involved in angiogenesis and in vascular smooth muscle cells contraction [4].
MMPs participate not only to the development of atherosclerosis, but also in its progression and in its complication: an increased MMP expression has been detected in atherosclerotic plaques and their activity may be responsible for plaque instability and rupture, and for an increased platelet aggregation [4].
Previously, we evaluated the behaviour of MMPs and TIMPs in metabolic syndrome and in obstructive sleep apnea syndrome, both conditions associated with arterial hypertension and with an elevated cardiovascular risk [17-21]. Considering that in the last decades several papers have focused on the role of MMPs and their inhibitors in arterial hypertension [22-26], our aim was to examine the literature data regarding their role in human hypertension and its complications, and the effects of some antihypertensive molecules on the MMP/ TIMP profile. Some authors have underlined that elevated levels of MMP-9 and TIMP-1 actually preceed the occurance of hypertension. By examining a population of participants to the Framingham Offspring Study, Dhingra et al have observed that subjects with higher TIMP-1 had more than double the possibility of having a hypertension occurance. In addition, there is a high risk to switch to a higher blood pressure classification, such as subjects with detectable concentration of MMP-9 [27].
MMPs and hypertension-induced vascular remodelling
Changes in blood pressure are responsible for different kinds of mechanical forces applied to the arterial wall: the thin wall hoop stress in the circumferential direction, the wall shear stress developed by blood flow and the axial stress induced by elongation in the axial direction. In response to these changes, the arterial vessels modify their thickness, composition and elastic properties [29].
Physiologically, vascular remodelling is an adaptive phenomenon of the circulatory system to changes in hemodynamic or metabolic demands, but chronically it contributes to atherosclerosis development and cardiovascular complications. The inward eutrophic remodelling is usually observed in resistance arteries in the early stages of hypertension, which is characterized by a reduction of the vessel lumen with normal media thickness and a rearrangement of vascular smooth muscle cells (VSMCs) [30, 31]. The renin-angiotensin system plays a pivotal role in vascular remodelling: angiotensin II has some effects on vascular fibroblast proliferation, ECM protein production and adhesion to matrix proteins. The hypertrophic remodelling is associated with an increase in the arterial wall thickness characterized by collagen deposition and elastin degradation, it is also common in large arteries, such as the aorta, in chronic severe hypertension [2].
MMPs play a specific role in blood vessel formation, remodelling and angiogenesis; moreover there are also some stimuli which induce MMPs expression and activation, such as oxidative stress, inflammation, angiotensin II, and hemodynamic forces [32] (Fig. 2).
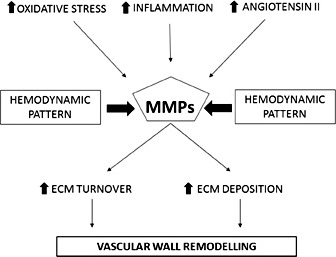
In the vascular wall of hypertensive subjects, there is an accelerated turnover and deposition of ECM components, mostly induced by MMPs. In addition, MMPs cleave large endothelin molecule producing smaller vasoactive fragments with a consequent activation of VSMCs and vasoconstriction [30]. Blood samples, of hypertensive subjects with atherosclerosis, show elevated levels of MMP-9 mRNA, whereas similar levels of TIMP-1 mRNA were found in comparison with normotensive or hypertensive subjects without atherosclerosis [33]. Tan et al demonstrated an increased concentration of MMP-9 and also of TIMP-1 in hypertensive subjects, positively correlated with carotid-femoral pulse wave velocity (PWV), but not with carotid-radial PWV [34]. This evidence suggested an implication of MMP-9 and its inhibitor in large arteries stiffness, but not in small muscular arterial elasticity [34]. However, MMP-9 polymorphisms are associated with higher MMP-9 levels, isolated systolic arterial hypertension [35] and carotid-femoral pulse wave velocity [36].
MMPs and hypertension-induced cardiac remodelling
Franz et al have examined hypertensive subjects with left ventricular hypertrophy observing higher levels of TIMP-1, TIMP-2 and TIMP-4 compared to controls, but also elevated MMP-9 levels, especially in concentric hypertrophy [37]. Saglam et al have demonstrated higher levels of MMP-3 and MMP-9 concentrations in hypertensive subjects with ventricular hypertrophy and a statistical correlation of these MMPs with left ventricular posterior wall thickness and Doppler indices of diastolic dysfunction [38]. Similarly, Ahmed et al have found an increased MMP-9 and TIMP-1 concentration in subjects with left ventricular hypertrophy and higher levels of TIMP-1 in those with chronic heart failure (CHF) [39]. TIMP-1 seems to be correlated to left ventricular mass and to the diastolic dysfunction degree (evaluated as mitral E/A ratio) [39, 40]. Recently, Ikonomidis et al have observed that the elevated levels of MMP-9 and TIMP-1 in hypertensive subjects are related to an impaired systolic myocardial deformation and to an increased arterial stiffness, evaluated as PWV [41]. Probably, an attenuation of the proteolytic activity of the MMPs, mediated by the elevated levels of tissue inhibitor, produces an accumulation of ECM proteins and, in particular, an increase in collagen content contributing to the development of the hypertensive heart disease (Fig. 3).
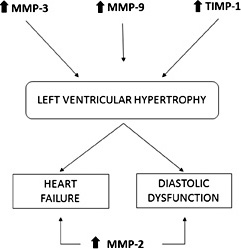
Some authors have suggested that an increase in MMP-9, TIMP-1, and in MMP-7 levels could predict the presence of left ventricular hypertrophy, while an increase in MMP-2 and a decrease in MMP-8 could predict the presence of diastolic heart failure [42]. Lately, the metanalysis of Marchesi et al has demonstrated elevated concentrations of MMP-9 and TIMP-1 in hypertensive subjects without heart failure and elevated levels of MMP-2 in hypertensives with heart failure, suggesting a role of the MMP-2 in left ventricular remodelling [28]. In animal models, active myocardial MMP-2 expression induces morphological alteration, such as myocyte hyperthrophy, myofilament lysis, sarcomeres destruction and cardiac fibroblast proliferation, resulting in ventricular remodelling [43].
In hypertensive subjects with paroxysmal atrial fibrillation, higher concentrations of TIMP-1 are correlated to an increased thickness of interventricular septum, to an increase in left ventricular mass index and to combined concentric and eccentric ventricular hypertrophy, which is prognostically unfavourable [44]. Even the TIMP-2 polymorphisms are significantly associated with the development of atrial fibrillation in subjects with hypertensive heart disease; a decreased expression of TIMP-2 may be responsible for an abnormal deposition of extracellular matrix, atrial fibrosis and structural remodelling promoting arrhythmias [45].
MMPs and antihypertensive molecules

ACE inhibitors and angiotensin receptor blockers
ACE inhibitors and ARBs show antioxidant effects [47-50]. Recently, several studies have examined the influence of these molecules on the MMP/TIMP profile in vivo, keeping in mind the results obtained in experimental models. In fact, in vitro both the ACE inhibitor temocaprilat and the ARB olmesartan inhibited not only the AGE-induced ROS generation but also the MMP-9 expression and activity in cultured smooth muscle cells and in monocytederived macrophages [51]. In samples of carotid artery from spontaneously hypertensive rats (SHR), a chronic treatment with a home-made ACE inhibitor reduced the expression of MMP-2 and MMP-9 and of inflammatory cytokines, such as IL-6 and TNF-α [52]. In the kidney of SHRs, the use of lisinopril or valsartan in monotherapy, or in combination, reduced the mRNA expression of TIMP-1 and MMP-9 [53]. In aortic VSMCs of Wistar rats, angiotensin II induced the expression of MMP-9, while losartan inhibited the expression of MMP-9 while enhancing that of TIMP-1 with a consequent decrease in MMP-9/TIMP-1 ratio in a concentration-dependent manner [54]. In addition, in SHR the renin inhibition with aliskiren reduced the myocardial expression of gelatinases and TIMP-1 after an ischemia/reperfusion injury attenuating the myocardial damage [55].
In a group of 35 hypertensive subjects, Fontana et al have noted no effects after a 8 week enalapril treatment on plasma levels and activity of MMP-2, MMP-9, MMP-8 and their inhibitors [56]. Derosa et al, in a cohort of subjects with arterial hypertension, have obtained a reduction in MMP-2 and MMP-9 concentrations after 13 months of losartan therapy, but the same result was not reached in ramipril-treated patients [57]. Differently, a 3 months period of anti-hypertensive treatment with lisinopril normalizes MMP-9/TIMP-1 ratio produced a reduction of plasma MMP-9 and an increase in TIMP-1 values, as well as in the use of candesartan [58].
The effects of ACE inhibitors and ARBs have been examined also in hypertensive subjects with cardiovascular complications. Schieffer et al have observed an improvement of MMP-9/TIMP-1 ratio in subjects with coronary artery disease treated with irbesartan or enalapril [59].
Hypertensive subjects with a high-degree of internal carotid artery stenosis, treated with irbesartan for 4 months, presented a reduced expression of MMP-2 and MMP-9 in the plaque areas of intense macrophage infiltration and a decreased inflammatory reaction [60]. The authors have suggested that the effect of irbesartan on plaque stabilization could be mediated by the inhibition of macrophages activity and the reduction of the oxidative stress. Other authors have demonstrated that a treatment with irbesartan reduced also the levels of collagenases MMP-1 and MMP-8 in subjects with high-degree carotid stenosis, but increased elastin degradation, indicating that the MMPs are not involved in elastin degradation of the atheroma [61].
The effects of ACE inhibitors and ARBs on MMPs levels seem to be mediated by different mechanisms: the inhibition of angiotensin II activity on MMPs expression, the antioxidant and anti-inflammatory effects. In addition, Yamamoto et al demonstrated that ACE inhibitors bind the active site of MMP-9 decreasing its activity [62, 63].
Calcium channel blockers (CCBs)
Animal models (rats) with renovascular hypertension treated with nifedipine, nimodipine, or amlodipine present a reduction of the oxidative stress and MMP-2 expression in thoracic aorta, inhibiting the vascular remodelling [64]. Similarly, a therapy with amlodipine and atorvastatin suppressed MMP-2 activity in the aorta of mice infused with angiotensin II for a period of 28 days [65]. It has been suggested that amlodipine decreases MMP-2 expression through the protein tyrosin kinase pathway [66]. Nifedipine and lercanidipine suppress the expression of inducible nitric oxide synthase mRNA, attenuate the TNF-α production and decrease the MMP-2, and MMP-9 expression and activity in VSMCs of rats treated with inflammatory stimuli [67]. In hypertensive rats, a treatment with nifedipine for 9 or 12 weeks reduced MMP-2 activity, left ventricular remodelling and cardiac dysfunction, independently of its antihypertensive action [68]. Lercanidipine promoted similar effects in diabetic rats [69]. In human macrophages treated with lacidipine for 24 hours, an evident decrease in MMP-9 and TIMP-1 expression has been observed [70].
In a group of hypertensive subjects, Martinez et al have demonstrated that the treatment with lercanidipine reduced MMP-9 plasma concentrations without affecting TIMP-1 or MMP-2 levels [71]. In 48 hypertensive subjects treated with olmesartan, the combination therapy with azelnidipine induced a reduction in MMP-2 levels and improved the arterial stiffness, differently from indapamide [72]. Azelnidipine seems to reduce the MMP-2 and MMP-9 gene expression as a pleiotropic antinflammatory effect, as it inhibits the macrophage infiltration into the vascular wall and the expression of proinflammatory cytokines [73]. Derosa et al have examined a group of 151 hypertensive subjects treated with losartan in combination with barnidipine or lercanidipine; both the CCBs have a slight reduction of MMP-2 and MMP-9 plasma levels, even if the decrease was not statistically evident [74]. However, Nakamura et al have demonstrated that telmisartan, but not amlodipine reduced MMP-9 levels in hypertensive subjects with chronic renal failure, suggesting that ARBs are more effective in this kind of patients [75].
Diuretics
Ceron et al [76] have studied the effects on animal models with renovascular hypertension, after a combined treatment with hydrochlorothiazide and spironolactone on MMP activity and vascular remodelling. They observed that the treatment with the two molecules, or the combination of the two, induced antioxidant effects and attenuated MMP-2 up-regulation. They suggested that the antioxidant action of the diuretics was due to the direct inhibition of vascular NADPH oxidase activity [76]. In animal models with hypertensive heart failure, eplerenone improved the cardiac remodelling decreasing the activity of MMP-2, MMP-7, MMP-12 and MMP-13 and regulating the expression of ECM proteins [77]. The data obtained from experimental studies demonstrate that aldosterone increases the expression of MMP-9 mRNA and the release of total and active MMP-9 proteins by human polymorphonuclear cells [78]. The actions of the aldosterone antagonists in humans have been evaluated mainly in subjects with chronic heart failure (CHF) than in hypertensive subjects. Li et al have demonstrated that spironolactone enhanced the cardiac function and significantly decreased plasma levels of MMP-1 and MMP-9 and the MMP-9/TIMP-1 ratio [79]. Similarly, Ogino et al have treated CHF subjects with spironolactone or furosemide for 16 weeks and observed a significant decrease in MMP-2 and MMP-9 levels only in the spironolactone-treated subgroup [80]. Derosa et al have evaluated the effects of canrenone on blood pressure and plasma biomarkers of vascular remodelling in subjects with metabolic syndrome; compared to a placebo treatment, canrenone significantly decreased blood pressure values, MMP-2 and MMP-9 concentrations [81].
Conclusion
Arterial hypertension is a recognized cause of atherosclerosis and cardiovascular events. The alterations in the composition and structure of the ECM are primary determinants in hypertension-induced vascular remodelling, these are largely mediated by an impairment in MMPs/TIMPs balance. On the contrary, the MMP inhibition mitigates the arterial remodelling of animal models with hypertension and atherosclerosis, as well as the arteral remodelling of humans. Therefore, a reasonable therapeutic strategy may be advantageous in hypertensive subjects in order to decrease the risk of cardiovascular complications.
Disclosure Statement
The authors declare that they have no conflict of interests.

